Results
[PDF]大環配位子結構與質子化系統及鑭系金屬離子錯合物結構之 ...
學生:林玉淳 ... 我們利用CHARMM 計算出來的能量來判斷配位子質子化位置的穩定 ... 另外,我們也利用量子化學軟體Gaussian 03 計算出鑭系金屬與多胺基多酸.
我們利用CHARMM 計算出來的能量來判斷配位子質子化位置的穩定性,能量越低表示越穩定。並可觀察到大 ... 另外,我們也利用量子化學軟體Gaussian 03 計算出鑭系金屬與多胺基多酸基大環配位子錯合後有配位水的最佳化結構。 ... 研究生: 林玉淳
[PDF]Chapter Ⅱ Literature review
https://ir.nctu.edu.tw/bitstream/11536/50713/7/850207.pdf
by 張峻愷 - 2003
Among them, the armchair CNTs have two integers n and m equal to each ... The chiral and zigzag nanotubes had two possibilities: (a) If n-m. = 3k, where k is a ...
28. 第三章. 計算化學理論方法簡介. 現今計算化學方法可依其所使用之理論背景,大致上約略可分為分子力學. (Molecular Mechanics) 、量子化學(Quantum ...
28. 第三章. 計算化學理論方法簡介. 現今計算化學方法可依其所使用之理論背景,大致上約略可分為分子力學. (Molecular Mechanics) 、量子化學(Quantum ...
第三章 計算化學理論方法簡介
現今計算化學方法可依其所使用之理論背景,大致上約略可分為分子力學
(Molecular Mechanics) 、量子化學 (Quantum Chemistry) 兩大宗。
以下就分子力學 (Molecular Mechanics) 、量子化學 (Quantum Chemistry)
理論方法作簡單介紹。
3.1 分子力學 (Molecular Mechanics, MM)
1930 年, Andrews 提出:在分子內,化學鍵都有“自然"的鍵長和鍵角值。
分子要調整它的幾何形狀 (geometry) ,使其鍵長和鍵角盡可能接近自然的值,
同時也要使非鍵結作用 (van der Waals) 處於最小的狀態,以給出原子核位置的
最佳分布;且在某些有張力的分子體系中,分子的張力也可以被計算出來。1946
年 Hill 提出利用 van der Waals 作用能和鍵長、鍵角的變形能來計算分子的能
量,以最適化分子的空間構型。 Hill 指出:分子內部的空間作用是眾所皆知的,
當基團或原子之間靠近時會相互排斥,為了減少這種作用力,基團或原子就趨於
相互離開,但是這將使鍵長伸長或鍵角發生彎曲,此又會引起相應的能量升高;
而最後的構型將會是此兩種力平衡後的結果,且是能量最低的構型。雖然分子力
學的思想和方法在 40 年代就建立起來了,但是直到 50 年代以後,隨著電腦的發
展,用分子力學來確定和理解分子的結構和性質之研究才越來越多。而最早的分
子力學 (molecular mechanics) 基本思想是 1930 年 由 Allinger 所提出,通常被 稱之為 MM 70, 71。
利用分子力學力場方法,輕微地改變分子的幾何結構,便能最佳化一個分子
單一構型的結構,並產生較合理的結構。分子的能量是可以利用一系列的數學函
數計算描述出分子的特徵,這些函數被稱之為位能函數 (potential energy
functions) 。分子被假定是有不同彈力的彈簧所結合之大量原子,分子的結構可
29
以藉由討論分子中一系列的點 (也就是原子) 與其所連結的彈簧 (也就是鍵) 之
間的關係,以得到此分子的描述與計算結果,而每一個彈簧就是一個平衡數值。
對提供彈簧一個外來的力,彈簧會從未平衡值回到平衡值,這就是所謂的虎克定
律 (Hooke's law) ;而這些位能函數的組合則是被使用於計算分子的總位能,此
即為力場 (force field)。
分子力學的計算方法是尋找系統的最低能量,即能量最小化過程。分子系統
有許多種能量極小狀態,而每個狀態對應特定能量,但總存在一個能量最小值,
也就是系統構型出現最大機率的能量。用分子力學的方法來描述分子在位能表面
上的運動狀態時,其總能量可由鍵長、鍵角、二面角、凡得瓦作用力、靜電力及
氫鍵等相關能量之總和來表示。
E (total) = E (stretching) + E (bending) + E (torsional) + E (van der Waals) + E
(electrostatic) + E (hydrogen bond) Eq.1
上式中 E(stretching ) 是鍵長伸縮的能量, E(bending) 是鍵角變化的能量,
E(torsional) 是二面角變化的能量, E(van der Waals) 是凡得瓦作用力,
E(electrostatic) 是靜電或偶極作用力,而 E(hydrogen bond) 為氫鍵的能量。其
各能量的貢獻詳細表示如下式:
E (total) = 2 0 ()L kLL −∑ + 2 0 ()k θ θ θ −∑ + [1 cos (| | )] i V n t + ⋅ Φ +∑ +
12 6 A B dd − + 12 123 2 1 3( )( ) [] || || R R DR R μμ μμ ⋅ ⋅ ⋅− uururuururuuruur ur + 12 10 332 DA DA HA HA q q AK BK d d d ε +−∑
Eq.2
當位能函數之形式及其參數決定後,即可根據分子內部各原子的位置算出整
30
個分子的總能量。
分子力場可根據量子力學的 Born-Oppenheimer Approximation,將原子核的
運動與電子的運動看成是獨立的。因此,一個分子的能量可近似為構成分子之各
個原子空間坐標的函數,亦即分子的能量隨分子構型之改變而變化,而描述這種
分子能量和分子結構之間的關係就是分子力場函數。分子力場函數來自於實驗結
果的經驗公式,相較於精確的量子力學全初始法,其對於分子能量的模擬是較粗
糙的,但分子力場方法的計算量較小數十倍,在適當的範圍內,分子力場方法的
計算精度與量子化學計算相差無幾,因此對於大分子複雜體系而言,分子力場方
法是一套可行的方法。
不同的分子力場會以不同形式的函數來描述上述方程式中能量與系統構型
之間的關係。至目前為止,不同的研究團隊設計了很多適用於不同系統的力場函
數,根據所選擇的函數及力場參數,可簡單分類為以下幾項:
1. 傳統力場
AMBER (Assisted Model Building and Energy Refinement) 力場:由
Kollman 團隊開發的力場,於生物大分子的模擬計算領域是被廣泛應用的一個分
子力場。而最初 AMBER 力場是專門為了計算蛋白質和核酸體系而開發的,計
算其力場參數的數據均來自實驗值,但隨著 AMBER 力場的廣泛應用,
AMBER 力場的內容不斷更新,逐漸開發出一個可以用於生物大分子、有機小分
子和高分子模擬計算的力場系統。總體來講, AMBER 力場的優勢在於生物大 分子的計算,其對小分子體系的計算結果常常不能令人滿意 72, 73。
CHARMM (Chemistry at HARvard Macromolecular Mechanics) 力場:由哈
佛大學 Karplus 團隊及其他研究團隊共同開發的分子力學計算工具,為一個被
廣泛承認並應用的程式,可以提供處理各種小分子、大分子 (包括蛋白質、核酸
和醣) 的經驗化能量計算,在其模擬過程可提供有關分子結構、相互作用、能量 等資訊。由小分子系統到溶劑化的大分子系統都有不錯的模擬結果 74-76。
CVFF (Consistent Valence Force Field) 力場: CVFF 力場原先的參數是用
31
於蛋白質結構的再現,最近幾年亦支持小分子的模擬,是一個可以用於無機系統 計算的力場,適合於含氫鍵的分子晶體 77。
MMX 力場: MMX 力場包括 MM 2 (the class 1 Allinger molecular mechanics program)70, 78 和 MM 3 (the class 2 Allinger molecular mechanics program)79,為目前應用最為廣泛的一種力場,主要針對有機小分子。
2. 第二代力場
第二代力場的位能函數形式比傳統力場更加複雜,涉及的力場參數更多,
計算量也更大,但也相對地更準確。
CFF (Consistent Force Field) 力場:由 Maple 團隊和 Hwang 團隊所開
發, CFF 為一個力場家族,由 CFF91 、 PCFF 、 CFF95 等許多力場所組成
而成。計算範圍涵蓋蛋白質、核酸、醣、脂類等生物大分子及有機小分子,特別 適用於複合物體系 (如蛋白質/DNA) 的相互作用研究 80。
COMPASS (Condensed-phase Optimized Molecular Potential for Atomistic Simulation Studies) 力場:由 MSI 公司 Sun 所開發出來的力場,為第一個基於
全初始計算所發展出來的力場,能夠準確預測孤立態和凝聚態分子的分子結構、 構型、振動及熱力學性質的分子力場,擅長進行高分子體系的計算 81。
3. 通用力場
通用力場所應用的力場參數是基於原子性質計算所得,使用者可以自主設
定一系列分子作為 training sets ,以生成所需的力場參數。
ESFF (extensible systematic forcefield) 力場:由 MSI 公司所開發的力
場,可以進行有機、無機分子的計算。 UFF (Universal Force Field) 力場:可計算周期表上所有元素的參數 82。 Dreiding 力場:適用於有機小分子、大分子、主族元素的計算 83。
而以分子力場為基礎的分子力學計算方法在分子動力學 (Molecular
Dynamics) 、蒙特卡羅 (Monte Carlo) 方法、分子嵌合 (Docking) 等分子模擬方
法中有著廣泛的應用。
32
3.1.1 分子動力學 (Molecular Dynamics, MD)70, 71, 84
在古典力學分子計算中,分子被視為一顆顆原子與一堆彈簧 (鍵結) 所組
成之彈簧組,其運動模式可用牛頓運動定律表示其兩原子間之位能關係。一分子
在定溫定壓經過一段時間後,各個原子的位移可透過牛頓運動定律,由位能函數
和合適的力場來得到。物體的移動可藉由計算短且不連續的時間間隔來完成,且
經由依次計算的速率以得知下一步之加速度,而初始速率是隨意地計算 (當由 0
K 開始,則動能必然會是 0) ,或藉由度量原子間初始的力來完成。模擬能在不
同溫度下執行,以獲得不同的構型物 (conformers),於高溫下,因較容易跨越過
能量障礙,故會得到更多的構形物。
分子動力學比分子力學所能得到的資訊更多更廣,分子動力學可用於研究
結構的穩定性,其任何與分子運動相關的性質皆可被計算出來,例如擴散、滲透、
分子振動模式、鍵結機制、隨時間的結構變動,以及利用其運動軌跡計算熱力學
性質等。
3.1.2 蒙地卡羅 (Monte Carlo) 法 71, 84
以機率和統計之理論及方法為基礎的一種計算方式,將所求解的問題用一
定的機率模型相聯繫,以電腦執行統計模擬或抽樣,以獲得問題的近似解,故又
稱統計模擬法。
蒙地卡羅 (Monte Carlo) 法能隨機地移動到一個新的幾何結構/構形,當新
分子構型能量低於原分子構型的能量,則接受新的構型,如果沒有,則會重新產
生一個新的構型,這個過程會一直持續到在某個特定次數產生一組低能量的構型
為止。
利用上述幾種模擬計算方法的計算軟體執行幾何結構最佳化 (geometry
optimization) 時,會在位能表面 (potential surface) 上找到與起始結構相近似的
33
分子平衡結構及能量,但由於能量極大值和能量極小值皆為位能表面上的靜止點
(stationary point) ,所以最佳化後的結果可能會收斂至能量最高或能量最低點,
且因分子的位能表面有很多個局部最低點 (local minimum) ,故通常要視情況而
再調整分子中的鍵長、鍵角、二面角和凡得瓦力等項目,以進一步找到所要的能
量最低點 (global minimum)。
分子力學和量子化學都是研究分子結構的理論方法,但二者存在著基本的差
別,分子力學方法可應用於包含數千個原子的大系統計算,且花費的成本與時間
比較少,與量子化學不同,分子力學一般較不考慮分子的電子結構,而是在半經
驗位能函數的基礎上直接處理分子中原子位置的問題。它的最大優點是計算速度
快。但是其限制為:
(1) 並不是只使用一種力場參數就能應用在所有分子系統上。倘若不同種類
的分子有其各自的力場描述,這樣才能得到較好的計算結果。
(2) 因忽略電子的影響,故不能用於探討以電子效應為主的的問題。例如無
法描述牽涉鍵的生成與斷裂的過程。
34
3.2 量子力學 (Quantum Chemistry) 法 85, 86
十八世紀末十九世紀初,量子力學開始發展。 1900 年 Planck 提出用常數
h (普朗克常數) 來表示相空間運動的一個量子, 1905 年 Einstein 發表了狹義
和廣義相對論的論文,使得現代關於時間和時間性質的想法產生突破性進展,並
給原子能的利用提供了理論基礎。直到 1926 年物理學家 Heisenberg 發表測不
準原理 (Uncertainty Principle) 及 1927 年物理學家 Schrödinger 發表薛丁格方
程式 (Schrödinger equation),奠定了量子力學的基礎。 1928 年 Heitler 與
London 完成了氫分子的量子計算後,科學家開始嘗試用量子力學的理論來解釋
化學物質的結構和化學現象。同年 Hartree 提出他的假設:將每個電子看成是在
其餘電子所提供的平均勢場中運動,利用迭代法 (iteration method ) 給出每一個
電子的運動方程式。 1930 年 Hartree 的學生 Fock 和 Slater 分別提出了考慮
庖立不相容原理 (Pauli’s exclusion principle) 的自洽場迭代方程和單行列式型多
電子體系波函數,此即現今的 Hartree-Fock 方程式,又稱為 HF 方程式, HF 方
程可以被稱為現代量子化學的基石。但是由於計算上的困難, HF 方程誕生後
沉寂了二十年。 1950 年,量子化學家 Roothaan 想到將分子軌道用原子軌道的
線性組合來近似展開,而得到閉殼層結構的 Roothaan 方程,發展出著名的 RHF
方程式,這個方程式及在這個方程基礎上做進一步發展的方法是現代量子化學處
理問題的根本方法。 1953 年美國的 Pariser 和英國的 Pople 花費兩年時間使用
手搖計算器分別達成對氮氣分子的 RHF 自洽場計算,這是人類首次藉由求解
HF 方程獲得對化學結構的量子力學解釋,也是量子化學計算方法第一次實際完
成。 1964 年物理學家 Kohn 指出:知道分佈在空間任意一點上的平均電子數
已經足夠,沒必要考慮每一個單電子的運動行為。這一思想帶來一種十分簡便的
計算方法--電子密度泛函理論 (Density Functional Theory,DFT) ,方法上的簡化
為大分子系統的研究帶來一線曙光;另外,密度泛函理論中還融入了統計的思
想,不必求每個電子的行為,只要求總電子密度,所以計算量大減,僅與電子數
35
成正比。化學家 Pople 並發展了化學中的許多計算方法,這些方法是基於對薛
丁格方程 (Schrödinger equation) 中的波函數作不同的描述,並建立了一套理論
模型化學 (Model Chemistry) ,採用 1950 年 Boys 提出用高斯函數展開的
STO 函數,用一系列越來越精確的近似值,促使量子化學方程的正確解析,進
而可以控制計算的精度。應用 Pople 的方法 (程式) ,把一個分子或一個化學反
應的特徵輸入電腦中,所得到的輸出結果就是該分子的性質或該化學反應可能如
何發生的具體描述,而這些計算結果可藉由通過 GAUSSIAN 程式顯示出來,能
被用於解釋或預測實驗結果。該程式的第一版本 GAUSSIAN 70 於 1970 年完
成,此後 Pople 和合作者相繼推出了從 GAUSSIAN 76 到 GAUSSIAN 03 九個
版本的程式系列。基於 Pople 的研究,從二十世紀 60 年代初開始建立了一系
列半經驗量子化學計算方法,包括有 NDDO (忽略雙原子微分重疊)、 CNDO (全
略微分重疊)、 INDO (間略微分重疊) 以及不同的參數化的 CNDO/1 、 CNDO/2
等方法,而後繼研究者繼續在 Pople 的研究基礎上發展出性能更好的半經驗量
子化學計算方法 MINDO/3 、 MNDO 、 AM1 、 PM3 、 ZINDO 和 SINDO1
等; Kohn 及 Pople 基於提出電子密度泛函理論及發展出普及的量子化學軟體
GAUSSIAN ,而獲得 1998 年諾貝爾化學獎。
3.2.1 全初始 (Ab inito) 法
一個分子的能量與性質在量子力學理論中,可用薛丁格方程式
(Schrödinger equation) 解出,但對於多電子原子或分子,由於其薛丁格方程式會
過於複雜,故無法求得其解。全初始法是使用數值求得與時間無關之薛丁格方程
式的近似解。
全初始法與分子力學方法或半經驗方法的差異在於在計算過程中不使用
任何來自實驗的參數,只使用以下幾個物理常數:光速,電子和核的電荷,及普
朗克常數,所有計算都建立在量子力學原理上,此即為為何這種方法被稱為 ab
36
inito 從頭算的原因。理論上使用全初始法所求得的計算結果會比較好,可較接
近正確值,但是因其所使用的電腦計算時間非常多,所以只適合於計算較小分
子。目前較常用之方法有: Hartree-Fock 及 MP2 等方法。
HF 方程式的基本概念為多電子體系波函數是由系統分子軌道波函數為
基礎所構造的 Slater 行列式 (Slater determinant) ,而系統分子軌道波函數則是
由系統中所有原子軌道波函數經過線性組合構成的,如果僅僅改變構成分子軌道
的原子軌道波函數係數,便能使系統能量達到最低點,而這一最低能量便是系統
電子總能量的近似,故在這一點上獲得的多電子體系波函數便是系統波函數的近
似。
全初始法與分子力學及半經驗法之不同點,主要是在於全初始法計算時不
需要用到實驗數據,而只是以量子力學為其依據來做計算,且全初始法可應用的
範圍相當廣泛,並不侷限於特定的系統,而早期的 ab inito 程式對於計算系統的
大小有很大的限制,但現今的 ab inito 程式已有很大的改善。
3.2.2 半經驗(Semi-empirical)法
半經驗法是一種電子結構的計算方法,所使用的理論基礎為量子力學,但
是在計算過程中大量的雙電子積分大部分是被省略,或是以參數代表,其所使用
的參數大都根據實驗數據,將一些波函數積分用經驗常數替代,如此可以減少上
千倍的計算量,基於採用的經驗常數不同,半經驗演算法的應用範圍也不同,故
應用時需要根據研究系統的具體情況進行選擇。
對於研究的化學系統,依據其可利用的適當參數去求得薛丁格方程式
(Schrödinger equation) 的近似解。常用的半經驗計算方法有: MNDO 、 AM1 、
MINDO/3 及 PM3 等方法。
半經驗法的限制為:
(1) 適用於組成原子的參數皆是詳細描述的系統,對於無適當參數可用
37
的原子,則無法得到較準確的計算結果。
(2) 關於氫鍵的描述、過渡狀態的結構或是缺乏足夠原子參數的分子而
言,半經驗法都無法求得良好的結果。
半經驗法的特點為:
(1) 可適用於非常大的分子系統;亦是唯一能實際使用量子力學方法於
大系統分子之計算方法。
(2) 可作為大系統分子計算的第一步。對於一個大系統分子的計算,可
先用半經驗法做最佳化 (optimization) ,先得到其最初的結構,進而再使用
Hartree-Fock 或電子密度泛函理論 (Density Functional Theory) 做最佳化。
3.2.3 電子密度泛函理論 (Density Functional Theory)
對於計算每個單電子運動描述的傳統分子性質,會使得在數學上的計算非
常複雜,因此, 1927 年 Thomas 和 Fermi 以電子密度為變數,利用統計力學
方法研究原子中電子的分佈,以電子密度來表示能量的理論,稱為 Thomas-Fermi
Model 。但由於這個理論模型有點粗糙,更嚴重的是用在化學方面,甚至出現
分子不會形成鍵結的計算結果,且因其完全沒有考慮到波函數的特點,故準確性
也不高,不過它的意義非常重要,因為此模型將電子動能第一次明確地以電子密
度形式表示,也就是以系統的電子密度為變數的方法。
1964 年, Kohn 及其助手和學生 Hohenberg 、 Sham 為理論化學帶來一
線曙光; Kohn 和 Hohenberg 證明了兩個非常重要的定理: Hohenberg-Kohn
第一定理告訴我們處於非簡併基態的多電子體系,其電子密度 r (r) 確定了該體
系的一切物理性質。例如該體系基態能量 E 0 可表示為 r (r) 的泛函,等於動
能、電子與核的作用勢能和電子與電子的作用勢能之和,之所以稱為“泛函",
是因為純量 E 0 是函數 r (r) 的函數。 Hohenberg-Kohn 第二定理相當於波函數
量子力學中的變分原理,由此可以嚴格證明用平均場觀念折算成單粒子的平均有
38
效勢能。將多體作用全部納入,最後可導出 Kohn-Sham 方程式,其計算量為
Hartree-Fock 水準,但卻已納入電子的交換和相關效應,計算精度優於
Hartree-Fock 方法,為量子化學的電子密度泛函理論奠定了基礎。
此後經過 Sham 等人二十餘年的努力, DFT 終於形成與分子軌道理論
並齊的嚴格的量子理論構架,它是用電子密度形式而不是波函數形式建成的另一
種形式的量子理論。此外, DFT 理論還把 Thomas-Fermi Model 和 Xα 方法都
統一在其範圍內。
電子密度泛函方程式理論的主要目的是利用電子密度代替大量電子和其
基本質量的波動方程式。在多個體波動方程式 (many-body wavefunction) 的計算
之中, N 個電子的波動方程式需要靠 3N 個變數來表示;相對於此來說,密度
是一個只有三個變數的量,故較簡單、較容易理解也較實用。如今,電子密度泛
函方程式已經成為量子化學中應用最廣泛的計算方法。
密度泛函理論 (Density Functional Theory,DFT) 理論本質上與
Hartree-Fock approximation 彼此獨立,而且所須計算量也差不多,但是密度泛函
理論是可以求出“精確解"的,不像 Hartree-Fock approximation 在一開頭就已
經取了近似。雖說是可以嚴格求解,但是在實用上卻仍難找出滿足精確解的
Hamiltonian ,此問題出在“交換關聯項“。所謂“交換關聯項“分成兩部份,
交換項是指 Hartree-Fock 位能項中不同種單電子波函數的交換積分,而關聯項
則是 Hartree-Fock 的動能項與真實動能項的差。
在全初始法之 Hartree-Fock 計算時,考慮的只是平均的電子密度,忽略了
電子的瞬間交互作用,而 DFT 方法則直接包含了電子交互作用的效應,因此
DFT 方法比起 Hartree-Fock 方法是增加了一些計算成本與時間,但是卻得到更
準確的計算結果。
對於分子的幾何結構、游離能等方面之探討,主要是採用全初始法或半經驗
法來做計算,而這些計算原理都是以量子力學為主,因此相當費時,故比較適合
39
於小分子的計算;對於中、大系統分子而言, DFT 方法所花費的成本少於
MP2 ,且能得到類似精確的計算結果;但若是較大的分子系統亦或在溶液系統
中之非反應性交互作用,則應以分子力學分法來計算較為適當,計算較為省時
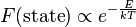
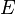 is state energy (which varies from state to state), and
is state energy (which varies from state to state), and 
是否可以搞成平面,看下水滴接触角啥的!
看接触角 和接触角迟滞,接触角越大越疏水,接触角迟滞越小越疏水
涂膜,滴水,然后看水的接触角
我这是粉末颗粒,可有方法...
我这是粉末颗粒,可有方法...
极性分子
编辑目录
分子概述编辑
化合价法
受力分析法
(2)若已知键角(或空间结构),可进行受力分析,合力为0者为非极性分子..如:CO2,C2H4,BF3
(3)同种原子组成的双原子分子都是非极性分子
有机极性判断编辑
偶极矩μ
分析1
分析2
结论
应用编辑
建议编辑
键与分子编辑