博文
玻璃形成能力的秘密  精选
精选
|||
金属玻璃作为一种新生代的金属材料,拥有很多新奇的物理、化学性能,比如说很好的化学催化反应活性(降解污水污染物、合成化学物质等)、优异的力学性能(高强度、高弹性、高硬度、耐磨等等),优异的软磁性能(饱和磁化强度高、矫顽力低,磁导率高等等),抗腐蚀能力强等。就是这样一种优异的金属材料吸引了越来越多的科学和工程工作者的注意力。那怎么样制备这种优异的金属材料呢?
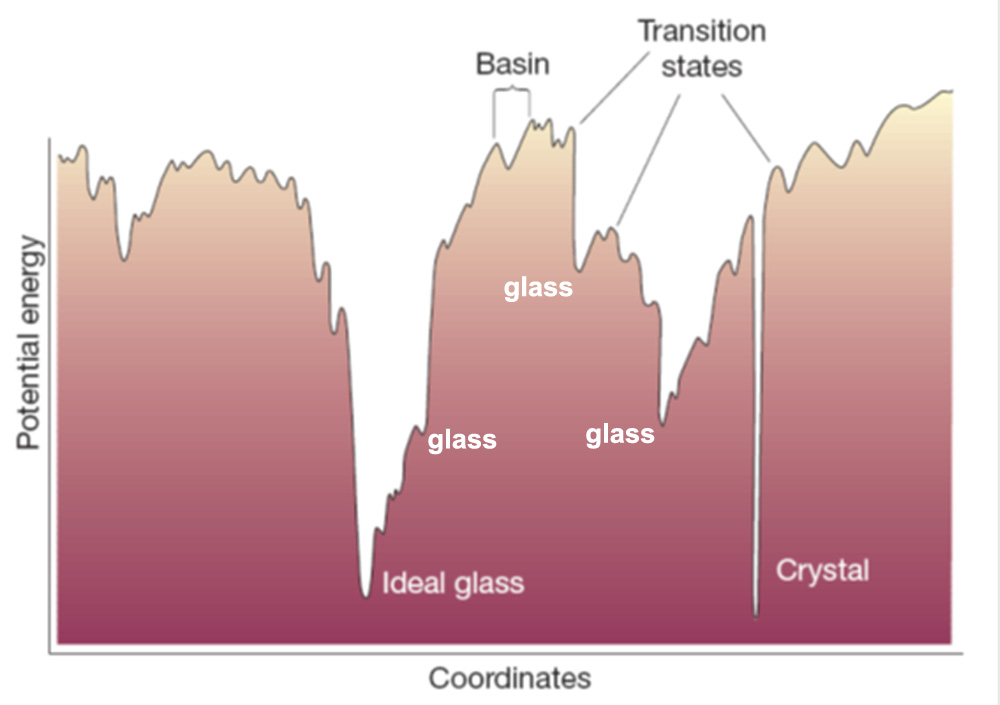
从能量势能图上我们可以看出晶态原子结构一般具有更低的能量,根据“水往低处流”的低能量更稳定的基本物理规律,很容易知道晶态是原子自发构筑时的首选结构;而无序的玻璃态原子结构的能量较高,更加不容易形成。因此制备金属玻璃需要抑制晶体的形成(形核、长大)。
一般制备金属玻璃的方法有以下几种:液体淬火法(最常见的制备方法)、气相沉积法、球磨法、辊轧法、离子辐射法等。这么多方法其实可以分为两大类:第一种就是通过抑制晶体的形成而得到金属玻璃,包括液体淬火法、气相沉积法等;第二种就是通过破坏晶体结构得到金属玻璃,包括球磨法、辊轧法、离子辐射法等。总之一句话,PK掉晶体,你就得到了玻璃。
因此,研究制备过程中晶体形成和玻璃形成的竞争关系对提高玻璃形成能力,控制玻璃的质量和性质有重要的意义。这也是从金属玻璃诞生半个世纪以来的永恒的话题,说白了就是研究热点。其实也是氧化物玻璃、有机玻璃、水溶液等玻璃研究领域的共同热点。而对于金属玻璃来说,研究玻璃形成能力的历史是思路比较清晰、框架比较明显的,这得益于金属玻璃属于玻璃家族中年轻的成员,之前的研究有了铺垫;金属合金体系的热力学框架基本建立起来了,包括相图、原子结构等;金属玻璃的原子结构比氧化物和有机物玻璃简单,等等历史原因。这些研究成果中主要包括以下几点,好的玻璃形成能力(低临界冷却速率)可以反映为:相图中的共晶点成分、高约化玻璃转变温度、宽过冷液相区、负混合焓等热力学判据,多组元成分、相异相似原子掺杂,自由电子分配原则等等。虽然金属玻璃才发现了50来年,但以上的这些理论相对来说还是比较古老了,因为都是上个世纪的产品。 金属玻璃之所以迷人,金属玻璃形成能力的话题之所以永恒,是因为人们总是能够时不时有一些激动人心的新的发现。进入二十一世纪以来,科学家们又有了很多突破性的发现。
说到这里不得不提一下金属玻璃家族的宠儿CuZr(铜锆)基金属玻璃体系,以CuZr二元合金为基础,大家做出了很多非常有影响力的工作,比如大玻璃形成能力体系、优异力学塑性体系,复合材料体系等等。可能从发表文章数量和引用数量来说,任何其他金属玻璃体系都无法与他相比。因此说他是金属玻璃研究领域的宠儿应该不为过。下面要介绍的两个关于玻璃形成能力的突破性成果也是围绕CuZr二元金属玻璃体系做出来的。
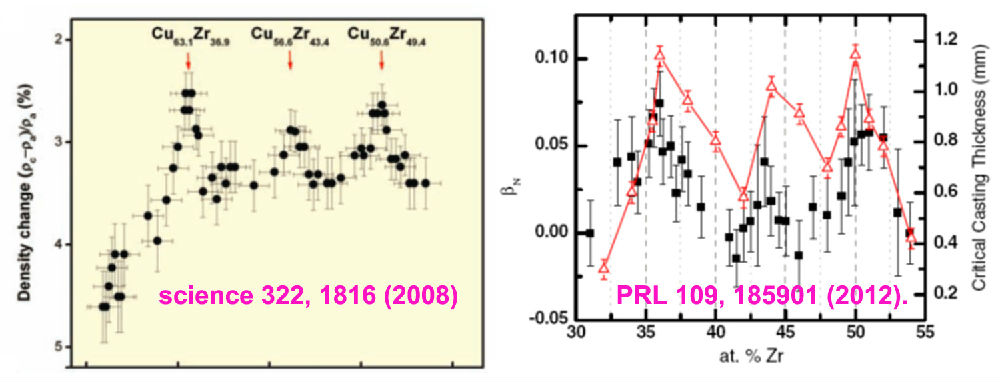
第一个要介绍的工作是2008年发表在Science 322, 1816(2008)上的文章,新加坡华裔科学家Li Y.教授与合作者发现,CuZr二元体系中,玻璃形成能力较好的成分点其原子堆积密度也比较大。因为原子在晶态中高度有序的密堆排列会使得密度更大,所以上面的结果让大家很自然的相信在金属玻璃中存在短程有序甚至中程有序原子堆积结构,对认识玻璃的本质及其形成能力有很大的帮助作用。而2012年在Phys.Rev.Lett. 109, 185901 (2012)上的文章,美国科学家Bendert和其合作者在同一个合金体系中进一步发现,这些玻璃形成能力好的体系之所以原子堆积比较紧密,不是因为其高温的液体中原子堆积紧密,而是因为其对应的液体在过冷降温过程中热膨胀(或叫冷收缩)系数比较大。这也和液体本身的脆度(fragility)有关系。这些都是非常漂亮、有启发性的工作。但是,如果你认为工作已经差不多完事了,那就大错特错了。
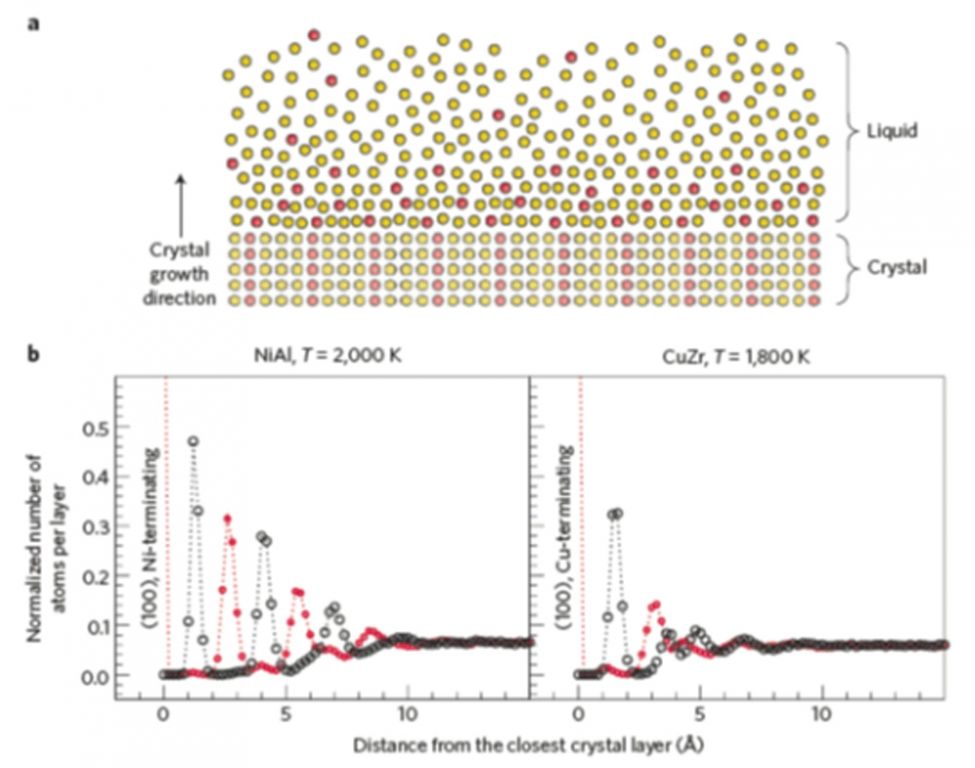
今年刚刚发表在Nature Mater. 12, 507 (2013)上文章,澳大利亚的Tang和Harrowell从更加直接的角度研究了CuZr体系的玻璃形成能力。上面提到了,玻璃形成的过程其实就是和晶体形成PK的过程。这个工作通过计算模拟在液体过冷降温过程中晶体在衬底表面形核、生长的行为。他们的结果表明,与玻璃形成能力较差的NiAl比较,CuZr金属液体在晶态衬底原子形成的周期势场作用较小,即从原子有序到原子无序的过度阶段较短。更形象的来说,在玻璃和晶体PK的战场上(晶态衬底和液体的界面处),玻璃态(具有类似液体的无序原子结构)更强的竞争力导致了CuZr比较好的玻璃形成能力。多么漂亮的工作!
玻璃形成,一个看似很简单的工业过程,里面包含了很多深刻的道理。很显然,关于玻璃形成能力的研究还远没有结束。路漫漫其修远兮,吾将上下而求索。你对这个感不感兴趣呢?
http://blog.sciencenet.cn/blog-202254-702149.html
上一篇:[转载]爱的诠释
下一篇:初见萤火虫
全部作者的其他最新博文
- • 祝大家圣诞快乐!
- • 科学网博客的bug
- • 大师做科研:上得了厅堂,下得了厨房
- • 非晶态物质的本质和特性
- • 我们只是需要一个想法!
- • 有一种失败叫做瞎忙
热门博文导读
17 严成 张雪峰 吕喆 田云川 吉宗祥 于全耀 胡瑞祥 张全成 郑玉峰 蒋敏强 樊晓英 李贵发 胡宝群 Rtqu cloud2 biofans ddsers
该博文允许注册用户评论 请点击登录 评论 (13 个评论)
- [13]张彦虎
- 年轻有为,

- 博主回复(2013-7-16 00:32):您是说文章中提到的这些人吧,呵呵
- [12]潘少鹏
- 一直在做结构,但是对于结构与GFA的关系还是稀里糊涂,没有什么思路
- 博主回复(2013-7-10 02:48):呵呵,上面的那篇PRL和NM说不定可以给你一些启发啊。
- [11]李贵发
- 军强哥分析得精到,
其实就是不同局域环境下的非晶团簇与晶核之间的竞争:如单个分子团簇,可以看做是真空环境下的形成能力比较;非晶形成过程,可以看作是液态环境凝固过程中,非晶团簇(五次对称)与晶核之间的竞争;非晶到晶体结构的高压转变形态,可以看做是固态环境下的非晶团簇或晶核之间的竞争。
正与学峰哥提的问题一样,环境因素太多,又像是到了蛇吃尾巴的自循环境遇。
金属玻璃真是其乐无穷啊!
- [10]张骥
- 对对。其实我们解释的也基本都是从你们实验室那里学过来的~这种现象应该比较普遍吧,贼纯贼纯的晶体应该挺少见的。我想知道有没有什么材料学晶体学方面的书或者文章有详细讲解这些过程的呢?或者有没有谁曾经拍到过这些晶体的生长过程呢?非常感谢你的回复哈!~学习了。
- [9]崔伟斌
- 博主是专家,强悍
顶一下 - 博主回复(2013-6-25 00:37):
 谬赞了
谬赞了
- [8]张骥
- 军强学长好!虽然我不是学材料的,不过主要的想法我也能够感觉到确实很巧妙精细。确实很有意思!~ 我是学矿物方面的,有个与晶体非晶体相关的问题想请教一下学长。就是一般天然矿物晶体生长中,总会有一些其他小的矿物晶体被包含在主晶里面,有没有什么晶体学方面的前辈研究过这些小晶体是如何包裹进入大晶体以至于长成我们现在看到的大晶体里面有小晶体的现象?我比较关心这个动力学过程。希望您能指教一下。谢谢!~
- 博主回复(2013-6-24 22:48):对地质矿物的演化过程真不清楚。不过实验室里这种现象应该挺多的,首先里面的小晶体可能是籽晶,触发了外面大晶体的形核和生长;另外还可能是两种成分的混合熔液在降温的过程中共同凝固结晶形成的。
- [7]mirrorliwei
- 有趣的话题。实际上能位图只是个静的指标,如同4楼所说的,反应速度也是个很重要的因子。因此,图1实质上应该是个3维的,要加一个反应速度的轴。另外参与反应的原子量也很重要。几个nm的厚度时成不了晶体,一过了临界厚度就会有相变的事态也会发生。
- 博主回复(2013-6-24 22:43):同意镜先生前面的看法
 。但是几个纳米厚度也可以是晶体,这个实验证据太多了。。。
。但是几个纳米厚度也可以是晶体,这个实验证据太多了。。。
- [6]于全耀
- 学习了,相对于博文内容,我更关心1楼和4楼的讨论。谢谢!
- 博主回复(2013-6-24 12:06):呵呵,雪峰兄发现了一个新现象。曙光,曙光,号外,号外~
- [5]崔伟斌
- 外行,说的不准确。抛砖引玉吧
- 博主回复(2013-6-24 11:20):谦虚了。
- [4]崔伟斌
- 回一楼:
热力学过程和动力学过程,都会导致非晶。
比如说,热力学上,晶态和非晶态的化学式差距不大,那么动力学上的成核时间差距不大,只要冷却不是准静态,很容易得到非晶。
如果热力学差距比较大,则动力学上的冷速的作用就比较重要了,冷慢了,就结晶了。
电沉积不是个准静态过程,类似于成核速率很高的过程,成核速率高,核尺寸小,小到一定程度就成非晶了。这个过程中,动力学因素的作用更大。
低温退火就晶化,很明显说明此态非稳态。
凝固是个动力和热力共同作用的过程 - 博主回复(2013-6-24 11:19):盛赞!

- [3]用户名
- 评论已经被科学网删除
- 博主回复(2013-6-24 12:08):赞~
- [2]张雪峰
- 太高深了
- 博主回复(2013-6-24 05:10):呵呵,你的实验搞得怎么样了?
- [1]张雪峰
- 在70多度溶液中化学镀Ni-P是非晶态,这意味着非晶态比晶态形成能更低、更稳定。但是通过简单的低温退火,非晶态又转变为晶体,这又说明晶态是更稳定的。军强能解释一下原因么?
- 博主回复(2013-6-24 02:47):另外,稳定与否是在平衡态或者是准静态情况下比较的。主要反映在那个体系的自由能更低。但是在玻璃形成的过程中是一个动态的过程,而玻璃形成和晶体形核是一个热力学涨落随机过程,改变外界条件可以改变其形成几率或喜好,所以才有了临界冷却速率。包括你这里镀膜用的溶液、衬底、镀膜速度等都应该是可调节因素。
- 博主回复(2013-6-24 02:39):突然想起你上次问我的问题了,原来答案在这里,呵呵
挺有意思的现象,定性来说,应该和衬底和镀膜原子之间的相互作用有很大关系,因为衬底的作用会限制其扩散。但是这种限制作用仅能局限在很小的范围内,包括晶格失配问题和化学势的扰动作用。这方面在很早的时候有人报道过电镀非晶膜,但是我不太了解。 而低温退火,就是弛豫作用了,因为NiP二元体系玻璃形成能力不好,稳定性较差,再加上镀的过程中有杂质和缺陷,会促进其晶化形核,从而很容易晶化。这个挖一挖应该有很多有意思的东西。
1/1 | 总计:13 | 首页 | 上一页 | 下一页 | 末页 | 跳转
|
化学键理论的发展
点击数:200 次 录入时间:2013/2/23 9:41:00 编辑:liuxinyuan2012 [宣传赚点]
世界上元素只有100多种,但目前已知化合物已超过1000万种了。元素是怎样形成化合物的,这是化学家共同关心的问题。
最早化学家假设原子和原子之间是用一个神秘的钩钩住的,这种设想至今仍留下痕迹,化学键的键字就有钩的意思。
1916年德国科学家柯塞尔考察大量的事实后得出结论:任何元素的原子都要使最外层满足8电子稳定结构。金属元素的原子易失去电子而成为带正电的阳离子,非金属元素的原子易获得电子而成为带负电的阴离子,从而各自达到稀有气体元素原子的最外层结构。形成的阴、阳离子靠库仑力结合成化合物。柯塞尔的理论能解释许多离子化合物的形成,但无法解释非离子型化合物。
1923年美国化学家路易斯发展了柯塞尔的理论,提出共价键的电子理论:两种元素的原子可以相互共用一对或多对电子,以便达到稀有气体原子的电子结构,这样形成的化学键叫做共价键。
科塞尔和路易斯的理论常叫原子价电子理论。它只能定性地描述分子的形成,化学家更需要对化学键作定量阐述。
1927年,海特勒(W.H.Heitler,1904——1981)和伦敦(F.London,1900-1954)用量子力学处理氢分子,用近似方法计算出氢分子体系的波函数和能量获得成功,这是用量子力学解决共价键问题的首例。1930年,鲍林更提出原子成键的杂化理论(杂化轨道理论)。1932年,洪德把单键、多键分成σ和π键两类。σ键指在沿着连结两个原子核的直线(对称轴)上电子云有最大重叠的共价键,这种键比较稳定。π键是沿电子云垂直于这条直线方向上结合而成的键,这种键比较活泼。这就使价键理论进一步系统化,使经典的化合价和化学键有机地结合在一起了。
由于上述的价键理论对共轭分子、氧气分子的顺磁性等事实不能有效解释,因此本世纪30年代后又产生一种新的理论——分子轨道理论。
分子轨道理论在1932年首先由美国化学家马利肯(R.S.Mulliken,1896——)提出来。他用的方法跟经典化学相距很远,一时不被化学界接受,后经密立根、洪德、休克尔、伦纳德等人努力,使分子轨道理论得到充实和完善。它把分子看作一个整体,原子化合成分子时,由原子轨道组合成分子轨道,原子的电子属于分子整体。分子轨道就是电子云占据的空间,它们可相互重叠成键。30年代后,美国化学家詹姆斯又使分子轨道理论计算程序化,能方便地用计算机处理,这便使分子轨道理论价值大大提高。接着美国化学家伍德沃德、霍夫曼发现分子轨道对称守恒原理和福田谦一等创立前沿轨道理论,使分子轨道理论大大地推前一步。
现代化学键理论已不只对若干化学现象作解释,而且理论已指导应用,如在寻找半导体材料、抗癌药物等方面起关键性作用。同时在90年代,现代价键理论已进入生命微观世界,从理论上认识酶、蛋白质、核酸等生命物质,从而进一步揭开生命的秘密。此外,近年来现代价键理论向动态发展,如化学反应进行中电子的变化情况,如何定量描述等。
总之,化学家对化学键的认识,从定性到定量,从简单到复杂,迄今可以说步步深入,估计本世纪中叶还有新的突破。
最早化学家假设原子和原子之间是用一个神秘的钩钩住的,这种设想至今仍留下痕迹,化学键的键字就有钩的意思。
1916年德国科学家柯塞尔考察大量的事实后得出结论:任何元素的原子都要使最外层满足8电子稳定结构。金属元素的原子易失去电子而成为带正电的阳离子,非金属元素的原子易获得电子而成为带负电的阴离子,从而各自达到稀有气体元素原子的最外层结构。形成的阴、阳离子靠库仑力结合成化合物。柯塞尔的理论能解释许多离子化合物的形成,但无法解释非离子型化合物。
1923年美国化学家路易斯发展了柯塞尔的理论,提出共价键的电子理论:两种元素的原子可以相互共用一对或多对电子,以便达到稀有气体原子的电子结构,这样形成的化学键叫做共价键。
科塞尔和路易斯的理论常叫原子价电子理论。它只能定性地描述分子的形成,化学家更需要对化学键作定量阐述。
1927年,海特勒(W.H.Heitler,1904——1981)和伦敦(F.London,1900-1954)用量子力学处理氢分子,用近似方法计算出氢分子体系的波函数和能量获得成功,这是用量子力学解决共价键问题的首例。1930年,鲍林更提出原子成键的杂化理论(杂化轨道理论)。1932年,洪德把单键、多键分成σ和π键两类。σ键指在沿着连结两个原子核的直线(对称轴)上电子云有最大重叠的共价键,这种键比较稳定。π键是沿电子云垂直于这条直线方向上结合而成的键,这种键比较活泼。这就使价键理论进一步系统化,使经典的化合价和化学键有机地结合在一起了。
由于上述的价键理论对共轭分子、氧气分子的顺磁性等事实不能有效解释,因此本世纪30年代后又产生一种新的理论——分子轨道理论。
分子轨道理论在1932年首先由美国化学家马利肯(R.S.Mulliken,1896——)提出来。他用的方法跟经典化学相距很远,一时不被化学界接受,后经密立根、洪德、休克尔、伦纳德等人努力,使分子轨道理论得到充实和完善。它把分子看作一个整体,原子化合成分子时,由原子轨道组合成分子轨道,原子的电子属于分子整体。分子轨道就是电子云占据的空间,它们可相互重叠成键。30年代后,美国化学家詹姆斯又使分子轨道理论计算程序化,能方便地用计算机处理,这便使分子轨道理论价值大大提高。接着美国化学家伍德沃德、霍夫曼发现分子轨道对称守恒原理和福田谦一等创立前沿轨道理论,使分子轨道理论大大地推前一步。
现代化学键理论已不只对若干化学现象作解释,而且理论已指导应用,如在寻找半导体材料、抗癌药物等方面起关键性作用。同时在90年代,现代价键理论已进入生命微观世界,从理论上认识酶、蛋白质、核酸等生命物质,从而进一步揭开生命的秘密。此外,近年来现代价键理论向动态发展,如化学反应进行中电子的变化情况,如何定量描述等。
总之,化学家对化学键的认识,从定性到定量,从简单到复杂,迄今可以说步步深入,估计本世纪中叶还有新的突破。
↓
原子形成分子有两种方式。一种是电子从一个原子完全转移到另一个原子上。失去电子的原子带正电,叫“正离子”,得到电子的原子带负电,叫“负离子”,它们通过正负电荷相互吸引靠在一起。比如氯化钠就是通过钠原子把一个电子完全交给氯原子而形成的。钠离子和氯离子之间这种联系叫做“离子键”,是“化学键”的一种。完全靠离子键形成的化合物一般是比较简单的无机物,比如盐类。
另一种方式不是电子转移,而是电子“共享”。你出一个电子,我出一个电子,这两个“共用电子”围绕两个原子核旋转,就把两个原子“栓”在一起了。这样形成的联系叫做“共价键”,生物大分子主要是靠共价键形成的。
不过共价键也有两种。一种是电子被两个原子“平均共享”,不偏向其中任何一个原子,两个原子都不带电。这种共价键就叫做“非极性键”,意思是化学键“两头”的电荷没有差别。比如碳原子和氢原子之间形成的化学键就是非极性键,所以完全由碳原子和氢原子组成的分子(叫“碳氢化合物”),无论是分子整体,还是局部,都没有固定的电荷,叫做“非极性分子”。
“共价键”的形成还有另外一种情形,是电子不被“平均共享”,而是偏向其中的一个原子。占有比较多电子的原子就带一些负电,占有比较少电子的原子就带一些正电,这样形成的化学键就叫做“极性键”。氧原子和氢原子之间共用电子形成的化学键就是“极性键”:共用电子偏向氧原子,使氧原子带一些负电,氢原子带一些正电。
氧原子“多占”电子的一个重要后果,就是水分子的奇特性质。水分子是由一个氧原子和两个氢原子共用电子形成的。这两个氢原子和氧原子并不在一条直线上,而是偏向氧原子的一边,两个化学键之间有104.5度的夹角。这样,水分子的正电荷中心和负电荷中心就彼此不重合,从总体上看就是水分子“一头”(氧原子“那头”)带负电,一头(两个氢原子“那头”)带正电,所以水分子是“极性分子”。
既然氧原子带负电,氢原子带正电,一个水分子中的氧原子就能够和其它水分子中的氢原子通过正负电荷而相互吸引,这样形成的联系叫做“氢键”。“氢键”的力量虽然没有“离子键”和“共价键”强,却是分子之间最强的作用力之一。水分子之间就是因为有“氢键”,彼此“抓”得很牢,所以水分子虽然很小(分子量只有18),水的沸点却很高(即水分子不容易“挣脱”其它水分子的吸引力,“飞”到空气中去),在一个大气压下水要到100摄氏度才“开锅”。而分子和水分子差不多大的“甲烷”(由1个碳原子和4个氢原子组成,分子量16),由于是“非极性分子”,沸点却低到零下161.5摄氏度,在常温常压下是气体。但是如果在甲烷分子中加一个氧原子,让它变成“甲醇”,沸点就增加到64.7摄氏度。一个氧原子和它形成的“氢键”竟然能使甲烷的沸点增加226.2摄氏度(161.5加64.7),说明氧原子“多占”电子所形成的分子内的“极性键”和分子之间的“氢键”在分子之间的相互作用上有多么大的作用!
“非极性分子”由于整体和局部都没有固定的电荷,按理说它们之间应该没有吸引力了,甲烷极低的沸点似乎也支持这个想法。但是汽油也是由许多不同的碳氢化合物的分子组成的,在室温下却是液体,说明分子之间有吸引力。由两个碘原子共用电子形成的碘分子也是“非极性分子”,因为这两个碘原子“旗鼓相当”,谁也别想“抢”谁的电子。按理说碘分子之间应该没有什么吸引力,但是提纯的碘却是固体,说明碘分子之间也有比较大的吸引力。这又该如何解释呢?
1930年,德裔美国科学家佛里茨·伦敦(Fritz London, 1900-1954)提出了一个假说来解释非极性分子之间的吸引力。他认为分子中电子的运动是动态的,虽然总体上看正电荷的中心和负电荷的中心彼此重合,但是在每一瞬间,这两个中心不一定完全重合,这就会产生瞬时的极性。这个极性又会影响相邻分子中电子的运动,在相邻的分子中“诱导”极性来,而且“诱导”出来的极性的方向与头一个分子中的极性方向相反(比如第一个分子中瞬时的局部负电荷会在相邻分子面向这个瞬时负电荷的地方“诱导”出正电荷来),这样两个分子就会相互吸引。通过这种机制形成的分子之间的吸引力叫做“伦敦力”。因为这种力不是固定在分子的某一部分的,而是随机发生在分子的大范围内,所以又称为“色散力”。
影响色散力大小的主要有两个因素,一个是原子和分子中电子瞬间“移位”的容易程度,二是分子之间接触面的大小。原子越大,里面的电子越多,电子就越容易瞬时“移位”。比如氟、氯、溴、碘是“同族”(位于“元素周期表”中同一竖行)的元素,外层电子结构相同,化学性质类似,也都由两个原子共用电子形成“非极性分子”。但是在常温常压下氟和氯是气体,溴是液体,而碘是固体。分子越大,里面的电子会越多,电子也更容易“移位”,分子之间的吸引力也会越强。比如由碳原子以单键线性相连,再连上氢原子形成的碳氢化合物(叫“正烷烃”)中,在常温常压下分子中有4个碳原子或以下的为气体(比如丙烷气),有5个到17个碳原子的为液体(汽油和煤油中的分子就在这个范围内),17个碳原子以上的为固体(比如石油蒸馏后留下的残渣)。分子之间的接触面越大,“诱导效应”就越容易发生,“色散力”也就越强。分子量相同的碳氢化合物中,分子的形状类似球形的,分子之间接触面就小,色散力就比较弱,而分子成线性的,分子之间的接触面大,色散力就比较强。比如同含5个碳原子的碳氢化合物“戊烷”中,碳链分支最多的“新戊烷”,沸点是9.5摄氏度,而碳链为直链的“正戊烷”,沸点是36.0摄氏度。
分子之间通过极性键(包括氢键)的相互作用,和通过色散力的相互作用,都是正电荷和负电荷之间的吸引,而且都只在短距离起作用(大约3到5个氢原子长度的范围内)。宇宙中的其它三种力,强作用力、弱作用力和万有引力,都与分子之间的作用无关。强作用力是把基本粒子(比如质子、中子和介子)结合在一起的力,作用范围比氢原子的尺寸还小100万倍。弱作用力和中子衰变为质子、电子和中微子有关,和分子之间的作用也没有关系。万有引力比电磁力弱1万亿亿亿亿倍,在分子的相互作用中可以完全忽略不计。所以分子之间的作用力,也是最后导致生命出现的力,只是电磁力。
极性键之间的作用力和色散力虽然都是电荷之间的作用力,它们之间却有重大差别。极性键中电荷是持续存在的,位置也是相对固定的,因此极性键之间的作用是“持续”和“定点”的,作用方式基本上是“点对点”。而色散力是随时变化的,电荷没有固定的位置,可以“平均”为分子之间的“大范围相互作用”,无法“精确定位”,作用方式是“面对面”,或者分子的“整体对整体”。在强度上,极性键之间的相互作用一般比色散力要强得多,除非“非极性分子”很大,接触面也很大。这两种作用方式不同的力彼此配合,在生物大分子的结构和细胞结构上起到关键的作用。
首先是各种分子在水中的溶解度。带有比较多“极性键”的分子,由于带有比较固定的电荷,能和水分子“亲密相处”,也就比较容易溶解在水中。这样的分子或分子局部就被称为是“亲水”的。比如葡萄糖的分子是由6个碳原子,6个氧原子和12个氢原子组成的,其中的6个氧原子带负电,而和它们相连的氢原子带正电,所以葡萄糖是高度溶于水的,每100毫升水可以溶解91克葡萄糖。而总体和局部都不带固定电荷的“非极性分子”,由于无法和水分子形成比较稳定的电荷相互作用,它们分散到水中时又会破坏水分子之间很强的相互作用,所以不受水分子的“欢迎”而被“排挤”出去,自己聚在一起,被称为是“憎水”的,也就是不溶于水。比如碳氢化合物“苯”(由6个碳原子连成环状,每个碳原子再连上一个氢原子所组成的化合物)就和水完全不混溶,所以是“憎水”的。但是苯却能够通过色散力和其它“非极性分子”相互作用,比如苯就可以溶解在汽油中。所以我们也可以把苯称为是“亲脂”的。
完全“亲脂”的分子(比如汽油中的分子)是不可能在水中形成固定结构的,因为它们在水中根本“呆不住”。完全“亲水”的大分子,即“全身”到处带电的分子,也不能在水中形成稳定的结构,因为它们的“身体”处处都受到水分子的包围,再加上水分子的热运动带来的冲击,没有一种力量能使它们稳定在一定的形状上。比如一种由葡萄糖单位线性相连组成的大分子叫做“直链淀粉”。它可以溶于热水,但是分子却没有固定的形状。要在水中形成稳定的立体结构,一个办法是分子上既有“亲水”的部分,又有“亲脂”的部分。“亲水”的部分可以处在结构表面,和水直接“打交道”,使分子或分子团能在水中稳定存在。而“亲脂”的部分由于受到水分子的排斥,被“赶”到一起,处于结构内部,从那里“拉住”分子的各个部分。这两种作用相互配合,就能在水中形成相对稳定的结构。
一个例子就是生物膜。要在水中形成生命,首要条件就是要把生命体系和周围的水环境分开来,这样组成生命的分子才不会被“稀释”和分散到水中去,不同生物体的遗传物质也不会相混,彼此干扰。所以最初的生命就必须采取“细胞”的形式,即有一个属于自己的,封闭的小空间,也就是所有的细胞都必须有自己的“墙壁”,这就是“细胞膜”。组成细胞膜的分子就是“两性”的,一头“亲水”,一头“亲脂”。当这样的分子被放到水中时,“亲脂”的部分被水“排挤”,彼此聚到一起,“亲水”的部分面向水,这样就能形成由两层分子组成的膜。每一层分子“亲脂”的部分都在膜内,彼此接触,但却不和水接触。每一层分子“亲水”的部分都朝向水,和水分子“亲密接触”。
许多两性分子都可以在水中形成双层膜。比如“脂肪酸”,它的“身体”主要是由碳原子和氢原子组成的长链,像汽油里面的分子,所以是高度“亲脂”的。和汽油里面的分子不同的是,脂肪酸的分子有一个比较“亲水”的,叫做“羧基”的“头部”(由一个碳原子上连上两个氧原子,其中一个氧原子再连上一个氢原子组成)。不过由脂肪酸组成的双层膜不是很“牢固”的,所以现在组成细胞膜的主要分子是“磷脂”。磷脂的分子组成比较复杂,是在甘油分子上连上两个脂肪酸和一个“磷酸根”,这个“磷酸根”再和一个“亲水”的分子(比如“丝氨酸”和“胆碱”)相连。所以磷脂也是“两性分子”,但是亲水和亲脂的部分都比较大。其中两根脂肪酸“尾巴”就是磷脂分子“亲脂”的部分,位于生物膜的内部。“磷酸根”和它所连的分子是高度“亲水”的,位于膜的外面,和水接触。
无论是细菌、植物、还是哺乳动物,细胞膜的构造都是由磷脂组成的双层膜,里面再“嵌镶”着一些蛋白质。这些细胞膜厚度相似,都在7-8纳米左右,中间的脂质层约厚2.5纳米,即大约有25个氢原子的厚度。如果检查组成细胞膜的磷脂里面的主要脂肪酸,发现它们都很长,比如棕榈酸和软脂酸有16个碳原子,油酸、亚油酸、亚麻酸和硬脂酸都有18个碳原子。这些脂肪酸都是高度不溶于水的,合成、吸收和运输都很麻烦,为什么生物要用这么长的脂肪酸呢?
主要原因估计有两个。一是细胞膜必须足够“结实”。细胞膜是细胞对外的“屏障”,容不得出任何差错。细胞膜破裂往往意味着细胞死亡。细胞膜除了要经受由周围分子的热运动造成的冲击(比如水分子的速度可以达到每秒694米,比波音飞机的速度还快3倍以上),而且还要耐受细胞内容物造成的渗透压(比如变形虫突然被雨滴击中)。而16到18碳原子长的脂肪酸才可以产生足够强的色散力,使碳氢链“尾巴”之间的作用力足够强。前面我们已经提到,17碳以上的烷烃,在常温常压下已经是固体。为了不让细胞膜真的成为“固体”,细胞膜已经采取了多种措施来保持其流动性,比如在膜中加入胆固醇,以及使用“不饱和脂肪酸”来“扰乱”脂肪层的结构。这意味着细胞已经把脂肪酸的长度推到形成“固体”的边缘,以求得足够的强度。
第二个原因是细胞膜必须成为离子的有效屏障。细胞内外的离子种类和数量的差别是很大的。比如细胞内有高浓度的钾离子和低浓度的钠离子;细胞外相反,有高浓度的钠离子和低浓度的钾离子。在细胞的“发电厂”线粒体中,内膜两边氢离子的浓度的差别也很大。这种膜两边离子浓度的差别对细胞的生理功能极为重要,所以膜必须防止离子“泄漏”。25个氢原子厚的脂质层对离子来讲就是脂肪的“汪洋大海”。即使是这样,轻度的“泄漏”仍在发生,要靠“离子泵”不断地把泄漏的离子“泵”回去。要是膜再薄,膜两边离子的浓度差就难以维持了。
另一个例子是细胞里面的遗传物质,脱氧核糖核酸(DNA)。大家都知道DNA的“双螺旋结构”,由“磷酸”和“核糖”(类似葡萄糖,但是只有5个碳原子)连成长链,核糖上连上“碱基”,碱基之间再通过氢键进行“配对”。其实“碱基”的作用不仅是“配对”。“碱基”,即腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶,是由碳原子和氮原子组成的单环(嘧啶)或双环(嘌呤)化合物,上面再连上其它原子或原子团。这些环由“共轭双键”(被单键隔开的双键)组成,分子是平面片状的。由于这些环结构中碳原子占一半以上,碳原子上面又连着氢原子,所以这些碱基的平面分子是比较“亲脂”的,这从它们在水中的低溶解度(除胞嘧啶的溶解度稍高以外,每升水只能溶1到2克)可以看出来。这些碱基分子的平面形状和“亲脂”性,使它们可以通过色散力紧密地“堆叠”在一起。理论计算表明,这种碱基之间的“堆叠效应”在维系DNA分子的结构上起主要作用。如果在碱基的环中加入氧原子,DNA美丽的双螺旋就无法存在了。
因此DNA和细胞膜一样,也是一个“夹心”结构。由磷酸和核糖组成的“亲水”链位于“双螺旋”的外边,与水“亲密接触”。“亲水”链的里面是由碱基“堆叠”成的两股“脂性螺旋”,在中心则是配对的“氢键”。所以DNA的结构也是由“亲水”和“亲脂”两种作用力相互配合来形成和维系的。
再一个例子是蛋白质。蛋白质是细胞中各种生理功能的“具体执行者”。它们不仅参与各种细胞结构的“建造”,还催化数以千计的化学反应。这些功能上的巨大要求使得蛋白质分子必须有各种特定的结构。蛋白质的这些结构又是如何形成和维持的呢?
蛋白质是由20种氨基酸按照一定的顺序线性相连形成的。但是像一根长线那样的蛋白质分子不仅没有生理功能,还容易彼此“缠”在一起,所以这些“长线”必须“卷”成一定的形状。详细叙述这个过程需要许多篇幅,但是简化了的图像也能说明问题。氨基酸,顾名思义,就是分子里面既有“氨基”,又有“酸基”,具体来说就是“羧基”。氨基和羧基能够彼此相连,这样就把氨基酸连成蛋白质了。除了氨基和羧基,氨基酸分子还有“侧链”,氨基酸连成“线”时,这些侧链就横向伸出,好像长线上横着伸出许多短线。这些侧链有些是“亲水”的(比如丝氨酸和谷氨酸),有的是亲脂的(比如亮氨酸和苯丙氨酸)。亲脂的侧链由于不受水分子“欢迎”,被“挤”到一起,位于分子的内部,亲水的侧链由于能与水分子相互作用,位于分子的外部,这样就把蛋白质的“长线”“卷”成“线球”了。根据亲脂和亲水氨基酸的排列顺序,就可以形成不同的蛋白质的分子结构,执行不同的功能。这些蛋白质分子的结构虽然千变万化,但是都是亲脂的侧链在内部,亲水的侧链在外部,蛋白质分子就像包了亲水“皮”的“油滴”。
前面讲了细胞膜是阻挡离子通过的屏障。但是细胞又需要和外界交换物质,包括各种离子和带电分子。这个功能也是由蛋白质来完成的。这些蛋白质分子必须“横穿”细胞膜,“沟通”膜的两边。这些蛋白质和溶解于水中的蛋白质不同,叫做“膜蛋白”。在这里蛋白质遇到了不同的环境:即有25个氢原子厚的“油层”。为了穿过这些“油层”,蛋白质分子有一个或多个区段,里面的侧链多数是亲脂的。这些亲脂节段可以容易地“穿过”细胞膜,而蛋白质中其余带有许多亲水侧链的节段则位于细胞膜之外。当一个膜蛋白有多个“穿膜节段”时(比如“离子通道蛋白”),这些“穿膜节段”也含有少数亲水的侧链。这些亲水侧链在脂性环境中被排斥,彼此通过固定电荷相互吸引,使这些“穿膜节段”彼此靠近,围成管状,形成“离子通道”。在这里蛋白质“穿膜节段”中亲水和亲脂侧链的位置就反过来了:亲脂的侧链朝外,与膜的脂性环境接触;亲水的侧链朝内,形成水性的通道。所以膜蛋白的结构也是由亲水和亲脂这两种作用力相互配合形成的,不过由于环境不同,“穿膜节段”的朝向和水溶性蛋白正好相反。
这里所举的只是几个最突出的例子,其中细胞膜的“夹层”结构,DNA“亲水双螺旋”里面的“亲脂双螺旋”,以及蛋白质在水中的“油滴结构”和在细胞膜中的“反油滴结构”,形成原理同出一辙,都是在水中时亲水部分包裹亲脂部分(“水包油”),在脂性环境中则是亲脂部分包裹亲水部分(“油包水”)。细胞的各种结构,比如细胞核、线粒体、内质网、溶酶体、肌纤维、纤毛、鞭毛,等等,其实也是主要靠亲水和亲脂这两种作用力以类似的方式彼此配合而形成和维持的。化学反应中“酶”和“底物”(被催化化学反应的物质)的相互作用,抗体和抗原的结合,“信号分子”和“受体蛋白质”的结合,也都涉及亲水和亲脂相互作用。由碳原子和氢原子对共用电子的“平等共享”导致的“亲脂”作用力,以及氧原子和其它原子(如氮原子)与氢原子共用电子时的“多吃多占”导致的“亲水”作用力,在各种化学结构的基础上,竟能“玩”出这么多“花样”来,让人觉得不可思议。生命过程虽然极其复杂,基本的作用力却相对“简单”。这让我们不能不惊叹生物进化过程对基本作用力加以利用的“本事”。

生物大分子和细胞怎样“组装”自己?
生物大分子和细胞怎样“组装”自己?——谈谈“亲水”和“亲脂”
生命是地球上最美妙的事物。郁郁葱葱的森林,一望无际的草原,叫腔婉转的鸟儿,翩翩起舞的蝴蝶,使这个世界充满生机。每一个生物体的构造又是那么完美精致,巧夺天工。哪怕是小小的蜻蜓,其构造的复杂程度和飞行性能都超过现代技术能造出来的任何飞行器。
在细胞和分子层面,生物一样能显现出它的精巧来。虽然多数细胞小到眼睛都看不见,但却是一个完整的“小世界”,里面有各种功能不同的“小房间”,有起支撑作用的“樑檩”,有自己的“指挥中心”、“发电厂”、“生产线”、“货物输送链”、“信息传递链”,“物品进出海关”、甚至有自己的“废品回收处理中心”。细胞如此,细胞里面的“生命大分子”,比如蛋白质和核酸,也都有非常规则的结构,并且在这些结构的基础上执行它们的生理功能。这么多复杂而规则的结构是怎么形成的呢?
地球上的生命是在水中形成的,现在的生物也都以水为介质。要问生物大分子和细胞的结构是如何形成的,其实就是在问这些结构是如何在水中形成的,这就要从原子如何形成分子说起。原子形成分子有两种方式。一种是电子从一个原子完全转移到另一个原子上。失去电子的原子带正电,叫“正离子”,得到电子的原子带负电,叫“负离子”,它们通过正负电荷相互吸引靠在一起。比如氯化钠就是通过钠原子把一个电子完全交给氯原子而形成的。钠离子和氯离子之间这种联系叫做“离子键”,是“化学键”的一种。完全靠离子键形成的化合物一般是比较简单的无机物,比如盐类。
另一种方式不是电子转移,而是电子“共享”。你出一个电子,我出一个电子,这两个“共用电子”围绕两个原子核旋转,就把两个原子“栓”在一起了。这样形成的联系叫做“共价键”,生物大分子主要是靠共价键形成的。
不过共价键也有两种。一种是电子被两个原子“平均共享”,不偏向其中任何一个原子,两个原子都不带电。这种共价键就叫做“非极性键”,意思是化学键“两头”的电荷没有差别。比如碳原子和氢原子之间形成的化学键就是非极性键,所以完全由碳原子和氢原子组成的分子(叫“碳氢化合物”),无论是分子整体,还是局部,都没有固定的电荷,叫做“非极性分子”。
“共价键”的形成还有另外一种情形,是电子不被“平均共享”,而是偏向其中的一个原子。占有比较多电子的原子就带一些负电,占有比较少电子的原子就带一些正电,这样形成的化学键就叫做“极性键”。氧原子和氢原子之间共用电子形成的化学键就是“极性键”:共用电子偏向氧原子,使氧原子带一些负电,氢原子带一些正电。
氧原子“多占”电子的一个重要后果,就是水分子的奇特性质。水分子是由一个氧原子和两个氢原子共用电子形成的。这两个氢原子和氧原子并不在一条直线上,而是偏向氧原子的一边,两个化学键之间有104.5度的夹角。这样,水分子的正电荷中心和负电荷中心就彼此不重合,从总体上看就是水分子“一头”(氧原子“那头”)带负电,一头(两个氢原子“那头”)带正电,所以水分子是“极性分子”。
既然氧原子带负电,氢原子带正电,一个水分子中的氧原子就能够和其它水分子中的氢原子通过正负电荷而相互吸引,这样形成的联系叫做“氢键”。“氢键”的力量虽然没有“离子键”和“共价键”强,却是分子之间最强的作用力之一。水分子之间就是因为有“氢键”,彼此“抓”得很牢,所以水分子虽然很小(分子量只有18),水的沸点却很高(即水分子不容易“挣脱”其它水分子的吸引力,“飞”到空气中去),在一个大气压下水要到100摄氏度才“开锅”。而分子和水分子差不多大的“甲烷”(由1个碳原子和4个氢原子组成,分子量16),由于是“非极性分子”,沸点却低到零下161.5摄氏度,在常温常压下是气体。但是如果在甲烷分子中加一个氧原子,让它变成“甲醇”,沸点就增加到64.7摄氏度。一个氧原子和它形成的“氢键”竟然能使甲烷的沸点增加226.2摄氏度(161.5加64.7),说明氧原子“多占”电子所形成的分子内的“极性键”和分子之间的“氢键”在分子之间的相互作用上有多么大的作用!
“非极性分子”由于整体和局部都没有固定的电荷,按理说它们之间应该没有吸引力了,甲烷极低的沸点似乎也支持这个想法。但是汽油也是由许多不同的碳氢化合物的分子组成的,在室温下却是液体,说明分子之间有吸引力。由两个碘原子共用电子形成的碘分子也是“非极性分子”,因为这两个碘原子“旗鼓相当”,谁也别想“抢”谁的电子。按理说碘分子之间应该没有什么吸引力,但是提纯的碘却是固体,说明碘分子之间也有比较大的吸引力。这又该如何解释呢?
1930年,德裔美国科学家佛里茨·伦敦(Fritz London, 1900-1954)提出了一个假说来解释非极性分子之间的吸引力。他认为分子中电子的运动是动态的,虽然总体上看正电荷的中心和负电荷的中心彼此重合,但是在每一瞬间,这两个中心不一定完全重合,这就会产生瞬时的极性。这个极性又会影响相邻分子中电子的运动,在相邻的分子中“诱导”极性来,而且“诱导”出来的极性的方向与头一个分子中的极性方向相反(比如第一个分子中瞬时的局部负电荷会在相邻分子面向这个瞬时负电荷的地方“诱导”出正电荷来),这样两个分子就会相互吸引。通过这种机制形成的分子之间的吸引力叫做“伦敦力”。因为这种力不是固定在分子的某一部分的,而是随机发生在分子的大范围内,所以又称为“色散力”。
影响色散力大小的主要有两个因素,一个是原子和分子中电子瞬间“移位”的容易程度,二是分子之间接触面的大小。原子越大,里面的电子越多,电子就越容易瞬时“移位”。比如氟、氯、溴、碘是“同族”(位于“元素周期表”中同一竖行)的元素,外层电子结构相同,化学性质类似,也都由两个原子共用电子形成“非极性分子”。但是在常温常压下氟和氯是气体,溴是液体,而碘是固体。分子越大,里面的电子会越多,电子也更容易“移位”,分子之间的吸引力也会越强。比如由碳原子以单键线性相连,再连上氢原子形成的碳氢化合物(叫“正烷烃”)中,在常温常压下分子中有4个碳原子或以下的为气体(比如丙烷气),有5个到17个碳原子的为液体(汽油和煤油中的分子就在这个范围内),17个碳原子以上的为固体(比如石油蒸馏后留下的残渣)。分子之间的接触面越大,“诱导效应”就越容易发生,“色散力”也就越强。分子量相同的碳氢化合物中,分子的形状类似球形的,分子之间接触面就小,色散力就比较弱,而分子成线性的,分子之间的接触面大,色散力就比较强。比如同含5个碳原子的碳氢化合物“戊烷”中,碳链分支最多的“新戊烷”,沸点是9.5摄氏度,而碳链为直链的“正戊烷”,沸点是36.0摄氏度。
分子之间通过极性键(包括氢键)的相互作用,和通过色散力的相互作用,都是正电荷和负电荷之间的吸引,而且都只在短距离起作用(大约3到5个氢原子长度的范围内)。宇宙中的其它三种力,强作用力、弱作用力和万有引力,都与分子之间的作用无关。强作用力是把基本粒子(比如质子、中子和介子)结合在一起的力,作用范围比氢原子的尺寸还小100万倍。弱作用力和中子衰变为质子、电子和中微子有关,和分子之间的作用也没有关系。万有引力比电磁力弱1万亿亿亿亿倍,在分子的相互作用中可以完全忽略不计。所以分子之间的作用力,也是最后导致生命出现的力,只是电磁力。
极性键之间的作用力和色散力虽然都是电荷之间的作用力,它们之间却有重大差别。极性键中电荷是持续存在的,位置也是相对固定的,因此极性键之间的作用是“持续”和“定点”的,作用方式基本上是“点对点”。而色散力是随时变化的,电荷没有固定的位置,可以“平均”为分子之间的“大范围相互作用”,无法“精确定位”,作用方式是“面对面”,或者分子的“整体对整体”。在强度上,极性键之间的相互作用一般比色散力要强得多,除非“非极性分子”很大,接触面也很大。这两种作用方式不同的力彼此配合,在生物大分子的结构和细胞结构上起到关键的作用。
首先是各种分子在水中的溶解度。带有比较多“极性键”的分子,由于带有比较固定的电荷,能和水分子“亲密相处”,也就比较容易溶解在水中。这样的分子或分子局部就被称为是“亲水”的。比如葡萄糖的分子是由6个碳原子,6个氧原子和12个氢原子组成的,其中的6个氧原子带负电,而和它们相连的氢原子带正电,所以葡萄糖是高度溶于水的,每100毫升水可以溶解91克葡萄糖。而总体和局部都不带固定电荷的“非极性分子”,由于无法和水分子形成比较稳定的电荷相互作用,它们分散到水中时又会破坏水分子之间很强的相互作用,所以不受水分子的“欢迎”而被“排挤”出去,自己聚在一起,被称为是“憎水”的,也就是不溶于水。比如碳氢化合物“苯”(由6个碳原子连成环状,每个碳原子再连上一个氢原子所组成的化合物)就和水完全不混溶,所以是“憎水”的。但是苯却能够通过色散力和其它“非极性分子”相互作用,比如苯就可以溶解在汽油中。所以我们也可以把苯称为是“亲脂”的。
完全“亲脂”的分子(比如汽油中的分子)是不可能在水中形成固定结构的,因为它们在水中根本“呆不住”。完全“亲水”的大分子,即“全身”到处带电的分子,也不能在水中形成稳定的结构,因为它们的“身体”处处都受到水分子的包围,再加上水分子的热运动带来的冲击,没有一种力量能使它们稳定在一定的形状上。比如一种由葡萄糖单位线性相连组成的大分子叫做“直链淀粉”。它可以溶于热水,但是分子却没有固定的形状。要在水中形成稳定的立体结构,一个办法是分子上既有“亲水”的部分,又有“亲脂”的部分。“亲水”的部分可以处在结构表面,和水直接“打交道”,使分子或分子团能在水中稳定存在。而“亲脂”的部分由于受到水分子的排斥,被“赶”到一起,处于结构内部,从那里“拉住”分子的各个部分。这两种作用相互配合,就能在水中形成相对稳定的结构。
一个例子就是生物膜。要在水中形成生命,首要条件就是要把生命体系和周围的水环境分开来,这样组成生命的分子才不会被“稀释”和分散到水中去,不同生物体的遗传物质也不会相混,彼此干扰。所以最初的生命就必须采取“细胞”的形式,即有一个属于自己的,封闭的小空间,也就是所有的细胞都必须有自己的“墙壁”,这就是“细胞膜”。组成细胞膜的分子就是“两性”的,一头“亲水”,一头“亲脂”。当这样的分子被放到水中时,“亲脂”的部分被水“排挤”,彼此聚到一起,“亲水”的部分面向水,这样就能形成由两层分子组成的膜。每一层分子“亲脂”的部分都在膜内,彼此接触,但却不和水接触。每一层分子“亲水”的部分都朝向水,和水分子“亲密接触”。
许多两性分子都可以在水中形成双层膜。比如“脂肪酸”,它的“身体”主要是由碳原子和氢原子组成的长链,像汽油里面的分子,所以是高度“亲脂”的。和汽油里面的分子不同的是,脂肪酸的分子有一个比较“亲水”的,叫做“羧基”的“头部”(由一个碳原子上连上两个氧原子,其中一个氧原子再连上一个氢原子组成)。不过由脂肪酸组成的双层膜不是很“牢固”的,所以现在组成细胞膜的主要分子是“磷脂”。磷脂的分子组成比较复杂,是在甘油分子上连上两个脂肪酸和一个“磷酸根”,这个“磷酸根”再和一个“亲水”的分子(比如“丝氨酸”和“胆碱”)相连。所以磷脂也是“两性分子”,但是亲水和亲脂的部分都比较大。其中两根脂肪酸“尾巴”就是磷脂分子“亲脂”的部分,位于生物膜的内部。“磷酸根”和它所连的分子是高度“亲水”的,位于膜的外面,和水接触。
无论是细菌、植物、还是哺乳动物,细胞膜的构造都是由磷脂组成的双层膜,里面再“嵌镶”着一些蛋白质。这些细胞膜厚度相似,都在7-8纳米左右,中间的脂质层约厚2.5纳米,即大约有25个氢原子的厚度。如果检查组成细胞膜的磷脂里面的主要脂肪酸,发现它们都很长,比如棕榈酸和软脂酸有16个碳原子,油酸、亚油酸、亚麻酸和硬脂酸都有18个碳原子。这些脂肪酸都是高度不溶于水的,合成、吸收和运输都很麻烦,为什么生物要用这么长的脂肪酸呢?
主要原因估计有两个。一是细胞膜必须足够“结实”。细胞膜是细胞对外的“屏障”,容不得出任何差错。细胞膜破裂往往意味着细胞死亡。细胞膜除了要经受由周围分子的热运动造成的冲击(比如水分子的速度可以达到每秒694米,比波音飞机的速度还快3倍以上),而且还要耐受细胞内容物造成的渗透压(比如变形虫突然被雨滴击中)。而16到18碳原子长的脂肪酸才可以产生足够强的色散力,使碳氢链“尾巴”之间的作用力足够强。前面我们已经提到,17碳以上的烷烃,在常温常压下已经是固体。为了不让细胞膜真的成为“固体”,细胞膜已经采取了多种措施来保持其流动性,比如在膜中加入胆固醇,以及使用“不饱和脂肪酸”来“扰乱”脂肪层的结构。这意味着细胞已经把脂肪酸的长度推到形成“固体”的边缘,以求得足够的强度。
第二个原因是细胞膜必须成为离子的有效屏障。细胞内外的离子种类和数量的差别是很大的。比如细胞内有高浓度的钾离子和低浓度的钠离子;细胞外相反,有高浓度的钠离子和低浓度的钾离子。在细胞的“发电厂”线粒体中,内膜两边氢离子的浓度的差别也很大。这种膜两边离子浓度的差别对细胞的生理功能极为重要,所以膜必须防止离子“泄漏”。25个氢原子厚的脂质层对离子来讲就是脂肪的“汪洋大海”。即使是这样,轻度的“泄漏”仍在发生,要靠“离子泵”不断地把泄漏的离子“泵”回去。要是膜再薄,膜两边离子的浓度差就难以维持了。
另一个例子是细胞里面的遗传物质,脱氧核糖核酸(DNA)。大家都知道DNA的“双螺旋结构”,由“磷酸”和“核糖”(类似葡萄糖,但是只有5个碳原子)连成长链,核糖上连上“碱基”,碱基之间再通过氢键进行“配对”。其实“碱基”的作用不仅是“配对”。“碱基”,即腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶,是由碳原子和氮原子组成的单环(嘧啶)或双环(嘌呤)化合物,上面再连上其它原子或原子团。这些环由“共轭双键”(被单键隔开的双键)组成,分子是平面片状的。由于这些环结构中碳原子占一半以上,碳原子上面又连着氢原子,所以这些碱基的平面分子是比较“亲脂”的,这从它们在水中的低溶解度(除胞嘧啶的溶解度稍高以外,每升水只能溶1到2克)可以看出来。这些碱基分子的平面形状和“亲脂”性,使它们可以通过色散力紧密地“堆叠”在一起。理论计算表明,这种碱基之间的“堆叠效应”在维系DNA分子的结构上起主要作用。如果在碱基的环中加入氧原子,DNA美丽的双螺旋就无法存在了。
因此DNA和细胞膜一样,也是一个“夹心”结构。由磷酸和核糖组成的“亲水”链位于“双螺旋”的外边,与水“亲密接触”。“亲水”链的里面是由碱基“堆叠”成的两股“脂性螺旋”,在中心则是配对的“氢键”。所以DNA的结构也是由“亲水”和“亲脂”两种作用力相互配合来形成和维系的。
再一个例子是蛋白质。蛋白质是细胞中各种生理功能的“具体执行者”。它们不仅参与各种细胞结构的“建造”,还催化数以千计的化学反应。这些功能上的巨大要求使得蛋白质分子必须有各种特定的结构。蛋白质的这些结构又是如何形成和维持的呢?
蛋白质是由20种氨基酸按照一定的顺序线性相连形成的。但是像一根长线那样的蛋白质分子不仅没有生理功能,还容易彼此“缠”在一起,所以这些“长线”必须“卷”成一定的形状。详细叙述这个过程需要许多篇幅,但是简化了的图像也能说明问题。氨基酸,顾名思义,就是分子里面既有“氨基”,又有“酸基”,具体来说就是“羧基”。氨基和羧基能够彼此相连,这样就把氨基酸连成蛋白质了。除了氨基和羧基,氨基酸分子还有“侧链”,氨基酸连成“线”时,这些侧链就横向伸出,好像长线上横着伸出许多短线。这些侧链有些是“亲水”的(比如丝氨酸和谷氨酸),有的是亲脂的(比如亮氨酸和苯丙氨酸)。亲脂的侧链由于不受水分子“欢迎”,被“挤”到一起,位于分子的内部,亲水的侧链由于能与水分子相互作用,位于分子的外部,这样就把蛋白质的“长线”“卷”成“线球”了。根据亲脂和亲水氨基酸的排列顺序,就可以形成不同的蛋白质的分子结构,执行不同的功能。这些蛋白质分子的结构虽然千变万化,但是都是亲脂的侧链在内部,亲水的侧链在外部,蛋白质分子就像包了亲水“皮”的“油滴”。
前面讲了细胞膜是阻挡离子通过的屏障。但是细胞又需要和外界交换物质,包括各种离子和带电分子。这个功能也是由蛋白质来完成的。这些蛋白质分子必须“横穿”细胞膜,“沟通”膜的两边。这些蛋白质和溶解于水中的蛋白质不同,叫做“膜蛋白”。在这里蛋白质遇到了不同的环境:即有25个氢原子厚的“油层”。为了穿过这些“油层”,蛋白质分子有一个或多个区段,里面的侧链多数是亲脂的。这些亲脂节段可以容易地“穿过”细胞膜,而蛋白质中其余带有许多亲水侧链的节段则位于细胞膜之外。当一个膜蛋白有多个“穿膜节段”时(比如“离子通道蛋白”),这些“穿膜节段”也含有少数亲水的侧链。这些亲水侧链在脂性环境中被排斥,彼此通过固定电荷相互吸引,使这些“穿膜节段”彼此靠近,围成管状,形成“离子通道”。在这里蛋白质“穿膜节段”中亲水和亲脂侧链的位置就反过来了:亲脂的侧链朝外,与膜的脂性环境接触;亲水的侧链朝内,形成水性的通道。所以膜蛋白的结构也是由亲水和亲脂这两种作用力相互配合形成的,不过由于环境不同,“穿膜节段”的朝向和水溶性蛋白正好相反。
这里所举的只是几个最突出的例子,其中细胞膜的“夹层”结构,DNA“亲水双螺旋”里面的“亲脂双螺旋”,以及蛋白质在水中的“油滴结构”和在细胞膜中的“反油滴结构”,形成原理同出一辙,都是在水中时亲水部分包裹亲脂部分(“水包油”),在脂性环境中则是亲脂部分包裹亲水部分(“油包水”)。细胞的各种结构,比如细胞核、线粒体、内质网、溶酶体、肌纤维、纤毛、鞭毛,等等,其实也是主要靠亲水和亲脂这两种作用力以类似的方式彼此配合而形成和维持的。化学反应中“酶”和“底物”(被催化化学反应的物质)的相互作用,抗体和抗原的结合,“信号分子”和“受体蛋白质”的结合,也都涉及亲水和亲脂相互作用。由碳原子和氢原子对共用电子的“平等共享”导致的“亲脂”作用力,以及氧原子和其它原子(如氮原子)与氢原子共用电子时的“多吃多占”导致的“亲水”作用力,在各种化学结构的基础上,竟能“玩”出这么多“花样”来,让人觉得不可思议。生命过程虽然极其复杂,基本的作用力却相对“简单”。这让我们不能不惊叹生物进化过程对基本作用力加以利用的“本事”。








 新浪微博
新浪微博 QQ
QQ 腾讯微博
腾讯微博 人人网
人人网
No comments:
Post a Comment