這是 Google 對 http://highscope.ch.ntu.edu.tw/wordpress/?cat=68 的快取。 這是該網頁於 2015年6月11日 17:34:20 GMT 顯示時的快照。 在此期間,目前網頁可能已經變更。 瞭解更多資訊
提示:如要在這個網頁上快速尋找您所搜尋的字詞,請按下 Ctrl+F 鍵或 ⌘-F 鍵 (Mac),然後使用尋找列進行搜尋。
提示:如要在這個網頁上快速尋找您所搜尋的字詞,請按下 Ctrl+F 鍵或 ⌘-F 鍵 (Mac),然後使用尋找列進行搜尋。
化學反應原理
反應式、化學反應式平衡、化學動力學、催化反應
中間體與穩定狀態近似法
中間體 (Intermediates)與穩定狀態近…
[新聞] 臺灣年輕團隊成功捕捉「克里奇中間體」與水分子的關鍵化學反應
臺灣年輕團隊突破研究氣壓上限 成功捕捉大氣活潑分子與水蒸氣之化學反應
中央研究院2015年1月6日新聞稿
大氣中一種名為「克里奇中間體」 (Criegee Intermediates,簡稱CIs,化學式R2COO,R為氫原子或烷基) 的活潑分子是大氣化學中的關鍵角色,更與大氣中的酸雨、懸浮微粒現象息息相關。然而由於CIs壽命十分短暫,很難捕捉或偵測,學界對此快閃神秘分子向來瞭解甚少。
中央研究院2015年1月6日新聞稿
大氣中一種名為「克里奇中間體」 (Criegee Intermediates,簡稱CIs,化學式R2COO,R為氫原子或烷基) 的活潑分子是大氣化學中的關鍵角色,更與大氣中的酸雨、懸浮微粒現象息息相關。然而由於CIs壽命十分短暫,很難捕捉或偵測,學界對此快閃神秘分子向來瞭解甚少。
{kind=link}
{kind=link}
{kind=link}
自由基(free radical)
自由基(free radical)
國立臺灣師範大學化學系碩士班二年郭修甫研究生
自由基(free radical),又稱做游離基,是一種半生期(half-life)非常短,形成後立即快速反應的反應中間體(intermediate),其成因大多是因為化合物分子在照射強光或加熱等條件下,共價鍵均勻性斷裂,而成為不具有成對電子的原子,也因為它有未成對的電子,所以他的反應性非常活躍而且極度不穩定,它必須從外部額外再取得一個電子,才能夠達到最穩定的狀態。
自由基的發現是因為摩西·岡伯格(Moses Gomberg)在利用三苯氯化甲烷(triphenyl methyl chloride)進行武茲偶聯反應(Wurtz reaction)想得到六苯乙烷(hexaphenylethane),但經過元素分析,他發現實際結果和算出來的理論值不相同,也發現這個產物,可以很快的和碘及氧氣進行反應形成三苯碘甲烷(triphenyl methyl iodide)和過氧化物(peroxide),因此他排除形成過氧化物的實驗條件,在二氧化碳中進行反應,但是結果反而出現岡氏二聚體的結構出現,經過反覆實驗結果,推測了自由基的結構(Figure 1)。
p1
國立臺灣師範大學化學系碩士班二年郭修甫研究生
自由基(free radical),又稱做游離基,是一種半生期(half-life)非常短,形成後立即快速反應的反應中間體(intermediate),其成因大多是因為化合物分子在照射強光或加熱等條件下,共價鍵均勻性斷裂,而成為不具有成對電子的原子,也因為它有未成對的電子,所以他的反應性非常活躍而且極度不穩定,它必須從外部額外再取得一個電子,才能夠達到最穩定的狀態。
自由基的發現是因為摩西·岡伯格(Moses Gomberg)在利用三苯氯化甲烷(triphenyl methyl chloride)進行武茲偶聯反應(Wurtz reaction)想得到六苯乙烷(hexaphenylethane),但經過元素分析,他發現實際結果和算出來的理論值不相同,也發現這個產物,可以很快的和碘及氧氣進行反應形成三苯碘甲烷(triphenyl methyl iodide)和過氧化物(peroxide),因此他排除形成過氧化物的實驗條件,在二氧化碳中進行反應,但是結果反而出現岡氏二聚體的結構出現,經過反覆實驗結果,推測了自由基的結構(Figure 1)。
p1
{kind=link}
烏龜再度打敗了兔子!─纖維素分解的關鍵因子
烏龜再度打敗了兔子!─纖維素分解的關鍵因子
國立臺灣大學生命科學系范姜文榮編譯/國立臺灣師範大學生命科學系李冠群副教授責任編輯
編譯來源:やはりカメはウサギに勝る
黴菌等絲狀真菌微生物,在自然界中以樹木或雜草為營養源,擔任分解者的角色。這些微生物能製造具有管狀構造的纖維素酶,它的特殊構造對分解結晶性纖維素相當重要。黴菌纖維素酶的管狀構造較其它絲狀真菌的纖維素酶為長,因此,兩者之纖維素酶的性質相異。但纖維素酶分解纖維素的反應,因為不溶性的結晶性纖維素與水溶性的產物形成固體與液體二相共存的「不均勻系」,實際反應作用分子與不反應分子同時混合在反應系統內,所以過去所使用的分解活性測定法,難以評估酵素分子的性質。
因此日本東京大學農學生命研究所與金沢大學的共同團隊,使用「高速原子力顯微鏡3」觀測各個酵素在分子層次上的運動模式。並研究黴菌Trichodarma reesei 所製造的纖維素酶TrCel7A、以及另一種真菌Phanerochaete chrysosporium 所製造的兩種纖維素酶 PcCel7C與 PcCel7D,藉由比較這3種酵素的分解反應速率和連續分解反應的次數,以解析影響結晶性纖維素分解的重要因子。
國立臺灣大學生命科學系范姜文榮編譯/國立臺灣師範大學生命科學系李冠群副教授責任編輯
編譯來源:やはりカメはウサギに勝る
S__311408
纖維素1是植物細胞壁的主要成分,是地球上現存最多的生物資源,因此世界各地許多研究者,正在研發將纖維素變換或製造成較易利用的酒精或有機酸。但纖維素具強鍵結的分子鏈,聚集成結晶構造,導致纖維素分解酵素-纖維素酶的分解效率低。{kind=link}
分解速度如「烏龜」般的纖維素酶,由於吸附性較強且能不斷進行分解反應,反而比如「兔子」般反應快速的纖維素酶更適合去分解結晶性纖維素。
黴菌等絲狀真菌微生物,在自然界中以樹木或雜草為營養源,擔任分解者的角色。這些微生物能製造具有管狀構造的纖維素酶,它的特殊構造對分解結晶性纖維素相當重要。黴菌纖維素酶的管狀構造較其它絲狀真菌的纖維素酶為長,因此,兩者之纖維素酶的性質相異。但纖維素酶分解纖維素的反應,因為不溶性的結晶性纖維素與水溶性的產物形成固體與液體二相共存的「不均勻系」,實際反應作用分子與不反應分子同時混合在反應系統內,所以過去所使用的分解活性測定法,難以評估酵素分子的性質。
因此日本東京大學農學生命研究所與金沢大學的共同團隊,使用「高速原子力顯微鏡3」觀測各個酵素在分子層次上的運動模式。並研究黴菌Trichodarma reesei 所製造的纖維素酶TrCel7A、以及另一種真菌Phanerochaete chrysosporium 所製造的兩種纖維素酶 PcCel7C與 PcCel7D,藉由比較這3種酵素的分解反應速率和連續分解反應的次數,以解析影響結晶性纖維素分解的重要因子。
追求不再貴重的催化之路
追求不再貴重的催化 (catalysis)之路
國立臺灣大學化學系名譽教授蔡蘊明
現代人類的生活和文明離不開化學,化學品的製造經常隨伴著副產物的產生,這些副產物若不謹慎處理,容易造成環境的污染。從效率的角度來看,產生無用而需廢棄的副產物,是一種浪費。以現在愈來愈受注重的綠色化學(註1)概念來看,我們需要發展更有效率的化學製程,其中催化劑的發展是一個重要的方向;催化劑可以降低化學反應的活化能,使得反應加速,因此能在較為溫和的條件下進行反應,明顯的具有節能的效果。催化劑扮演的是協助的角色,本身並不會成為產物的一部分;透過一個循環的機制,催化劑在每一次的循環結束時,會重新產生,進行下一輪的催化循環,因此並不需要使用許多的催化劑。在工業上,好的催化劑用量最好能在0.01當量以下,愈少愈好,若是超過0.05當量,將不會是很理想的催化劑。
國立臺灣大學化學系名譽教授蔡蘊明
現代人類的生活和文明離不開化學,化學品的製造經常隨伴著副產物的產生,這些副產物若不謹慎處理,容易造成環境的污染。從效率的角度來看,產生無用而需廢棄的副產物,是一種浪費。以現在愈來愈受注重的綠色化學(註1)概念來看,我們需要發展更有效率的化學製程,其中催化劑的發展是一個重要的方向;催化劑可以降低化學反應的活化能,使得反應加速,因此能在較為溫和的條件下進行反應,明顯的具有節能的效果。催化劑扮演的是協助的角色,本身並不會成為產物的一部分;透過一個循環的機制,催化劑在每一次的循環結束時,會重新產生,進行下一輪的催化循環,因此並不需要使用許多的催化劑。在工業上,好的催化劑用量最好能在0.01當量以下,愈少愈好,若是超過0.05當量,將不會是很理想的催化劑。
催化劑簡介
催化劑基本上分為兩種:異相催化與勻相催化。前者是使用的催化劑與反應溶液不互溶,催化的反應發生在催化劑與溶液的介面,因此催化劑的表面積愈大效果愈好。常見的異相催化劑,例如食品工業中,將不飽和脂肪酸的碳-碳雙鍵飽和化時,使用氫氣為還原劑,但需要鈀(Pd)、鉑(Pt)或銠(Rh)等金屬做為催化劑。這些金屬不溶於反應使用的有機溶劑,屬於異相催化劑。為了充份將金屬的表面攤開以提高效率,細微的金屬顆粒是靠著吸附的方式附著在各種固相的擔體上面,常用的擔體是木炭(charcoal)的粉末。異相催化的好處是去除容易,透過簡單的過濾即可,需要的話可以回收再使用,符合綠色化學的精神;但壞處是表面積的多寡與顆粒的大小和均勻度有關,不易控制。反應發生在兩相介面,攪拌的效率很重要,因此反應的時間必須視實際進行狀況來判定。不同的擔體會影響金屬的表面結構(註2),進而影響反應活性和選擇性,不同的金屬化學反應性也不同。
a1
勻相催化的系統中,催化劑是可溶於反應溶劑中的,因此反應是發生在均勻的單相中,反應的速率易於掌控,但是去除催化劑以及回收不易。許多的勻相催化劑乃以金屬為催化的核心,那麼要如何讓金屬溶於有機溶劑呢?操控溶解度是利用金屬的配位能力,使用有機化合物做為配位基(ligand),與金屬生成的配位化合物(coordination compound),被稱為有機金屬化合物(organometallic compound),可以溶於有機溶劑 (圖1)。這些有機金屬的催化劑另一項最重要的優點,在於有機配位基的結構可以改變,進而影響金屬催化的活性以及選擇性,這就給予了化學家很大的空間去發展符合他們需求的催化劑。{kind=link}
圖1 銠(Rh)金屬是碳-碳雙鍵氫化常用的異相催化金屬,將三氯化銠與過量的三苯磷反應可得到著名的威爾金森催化劑(Wilkinson catalyst),此催化劑可以溶解於許多的有機溶劑中。
金催化反應(Gold Catalyzed Reactions)(I)
金催化反應(Gold Catalyzed Reactions)(I)
國立臺灣師範大學化學系梁家榮博士班二年級
黃金是人類史上相當早就發現的金屬之一,西元前2500年前在埃及的雕刻上,就可以發現類似開採或冶煉的圖像。一般認為黃金是惰性的、極不易被氧化且不具催化活性的。由於黃金在地殼中的豐度約為 5×10-7%,屬於貴重金屬,故其價格也非常昂貴。尤其在近年黃金的市場價格大幅上漲,在過去幾千年的歷史中,僅僅用於做為貨幣或飾品的功用居多1。後來發現金具有良好的延展性且為抗腐蝕良導體的性質,才開始在電子產品上有更多應用。黃金與大多數化學試劑不發生反應,但是可以溶於王水中,並可以再進一步製備而得到金(I)和金(III)試劑。近年來發現金(I)和金(III)離子在勻相催化( homogeneous catalysis)中對帶有炔基(alkyl group)的化合物具有催化活性之後,過去十幾年間世界各地的化學實驗室競相報導關於金催化的研究,掀起一波新的掏金熱潮。
國立臺灣師範大學化學系梁家榮博士班二年級
黃金是人類史上相當早就發現的金屬之一,西元前2500年前在埃及的雕刻上,就可以發現類似開採或冶煉的圖像。一般認為黃金是惰性的、極不易被氧化且不具催化活性的。由於黃金在地殼中的豐度約為 5×10-7%,屬於貴重金屬,故其價格也非常昂貴。尤其在近年黃金的市場價格大幅上漲,在過去幾千年的歷史中,僅僅用於做為貨幣或飾品的功用居多1。後來發現金具有良好的延展性且為抗腐蝕良導體的性質,才開始在電子產品上有更多應用。黃金與大多數化學試劑不發生反應,但是可以溶於王水中,並可以再進一步製備而得到金(I)和金(III)試劑。近年來發現金(I)和金(III)離子在勻相催化( homogeneous catalysis)中對帶有炔基(alkyl group)的化合物具有催化活性之後,過去十幾年間世界各地的化學實驗室競相報導關於金催化的研究,掀起一波新的掏金熱潮。
金催化反應(Gold Catalyzed Reactions)(II)
金催化反應(Gold Catalyzed Reactions)(II)
國立臺灣師範大學化學系梁家榮博士班二年級
請參考連結:「金催化反應(Gold Catalyzed Reactions)(I)」
金離子相對於銀離子具有更多一層軌域可以提供配位(coordinate),以帶有炔基的反應物為例(如圖一)1,金催化的反應機制(mechanism)首先是先由富含有π電子的炔基提供電子配位到金(I)離子上。而圖中銀試劑的功用是先與鹵素反應生成難溶鹽類的沈澱物氯化銀,此時催化劑屬於sp混成軌域,結構為直線型的金(I)離子就能空出一個可配位的空間,成為缺電子的路易士酸能與炔基進行反應。經由金(I)離子活化的炔基會非常容易與親核試劑(nucleophile)行反式加成(anti-addition)反應,也就是金離子會和親核試劑處在反邊的位置上。最後反應可能透過金離子上的電子共振形成金碳烯(carbene)的碳金雙鍵結構,這種結構可以在低溫條件下存在,也已經透過X-ray 繞射分析(X-ray diffraction)證實。2
國立臺灣師範大學化學系梁家榮博士班二年級
請參考連結:「金催化反應(Gold Catalyzed Reactions)(I)」
金離子相對於銀離子具有更多一層軌域可以提供配位(coordinate),以帶有炔基的反應物為例(如圖一)1,金催化的反應機制(mechanism)首先是先由富含有π電子的炔基提供電子配位到金(I)離子上。而圖中銀試劑的功用是先與鹵素反應生成難溶鹽類的沈澱物氯化銀,此時催化劑屬於sp混成軌域,結構為直線型的金(I)離子就能空出一個可配位的空間,成為缺電子的路易士酸能與炔基進行反應。經由金(I)離子活化的炔基會非常容易與親核試劑(nucleophile)行反式加成(anti-addition)反應,也就是金離子會和親核試劑處在反邊的位置上。最後反應可能透過金離子上的電子共振形成金碳烯(carbene)的碳金雙鍵結構,這種結構可以在低溫條件下存在,也已經透過X-ray 繞射分析(X-ray diffraction)證實。2
金(I)離子的催化反應圖。
{kind=link}
圖一、 金(I)離子的催化反應圖。
配位錯合物(Coordination Complex) (II)
配位錯合物(Coordination Complex) (II)
國立臺灣師範大學附屬中學二年級1322班楊易蓁/國立臺灣師範大學附屬高級中學化學科陳昭錦老師
1.氧化還原反應:
配位錯合物中的氧化還原反應可歸納為內圈(inner sphere)及外圈(outer sphere)電子轉移兩種機制。內圈電子轉移的機制是由1983年諾貝爾化學獎得主亨利陶比(Henry Taub)提出的,比較方程式(1)及(2):
[Co(NH3)6]3+ + [Cr(H2O)6]2+ → [Co(NH3)5(H2O)]2+ + [Cr(H2O)6]3+ (1)
[CoCl(NH3)5]2+ + [Cr(H2O)6]2+ → [Co(NH3)5(H2O)]2+ + [CrCl(H2O)5]2+ (2)
在方程式(1)中,[Co(NH3)6]3+的中心原子Co由+3變為+2,[Cr(H2O)6]2+ 的中心原子Cr由+2變為+3。而在方程式(2)中當Co的其中一個配位基改為Cl-時,當發生電子轉移時,Cl-也從原本與氧化劑中的Co2+配位變成與還原劑中的Cr3+配位。此外,方程式(2)的反應速率也比(1)快很多。
陶比認為在方程式(2)中,Cl-扮演電子流的導管,使其能從Cr2+轉移到Co3+,形成Cr3+與Co2+。Cl-扮演兩個中心原子的配位基,形成一雙金屬錯合物的中間體[Co(NH3)5(μ-Cl)Cr(H2O)5]4+,作為電子傳遞的媒介,其中"μ-Cl" 意指Cl-擔任Cr及Co原子的橋鍵配位基(bridging ligand)。透過內圈電子轉移的機制,電子可以由一個配位化合物的中心轉移至另一個。
外圈電子轉移的機制則是參與氧化還原反應的中心原子並未藉由任何橋樑進行電子轉移,而是電子從還原劑的中心”跳躍”(hop)至氧化劑,過程中配位基並未發生任何變化,參見圖一的例子。
國立臺灣師範大學附屬中學二年級1322班楊易蓁/國立臺灣師範大學附屬高級中學化學科陳昭錦老師
配位錯合物的化學反應
配位錯合物的化學反應主要有氧化還原反應及配位基取代兩大類:1.氧化還原反應:
配位錯合物中的氧化還原反應可歸納為內圈(inner sphere)及外圈(outer sphere)電子轉移兩種機制。內圈電子轉移的機制是由1983年諾貝爾化學獎得主亨利陶比(Henry Taub)提出的,比較方程式(1)及(2):
[Co(NH3)6]3+ + [Cr(H2O)6]2+ → [Co(NH3)5(H2O)]2+ + [Cr(H2O)6]3+ (1)
[CoCl(NH3)5]2+ + [Cr(H2O)6]2+ → [Co(NH3)5(H2O)]2+ + [CrCl(H2O)5]2+ (2)
在方程式(1)中,[Co(NH3)6]3+的中心原子Co由+3變為+2,[Cr(H2O)6]2+ 的中心原子Cr由+2變為+3。而在方程式(2)中當Co的其中一個配位基改為Cl-時,當發生電子轉移時,Cl-也從原本與氧化劑中的Co2+配位變成與還原劑中的Cr3+配位。此外,方程式(2)的反應速率也比(1)快很多。
陶比認為在方程式(2)中,Cl-扮演電子流的導管,使其能從Cr2+轉移到Co3+,形成Cr3+與Co2+。Cl-扮演兩個中心原子的配位基,形成一雙金屬錯合物的中間體[Co(NH3)5(μ-Cl)Cr(H2O)5]4+,作為電子傳遞的媒介,其中"μ-Cl" 意指Cl-擔任Cr及Co原子的橋鍵配位基(bridging ligand)。透過內圈電子轉移的機制,電子可以由一個配位化合物的中心轉移至另一個。
外圈電子轉移的機制則是參與氧化還原反應的中心原子並未藉由任何橋樑進行電子轉移,而是電子從還原劑的中心”跳躍”(hop)至氧化劑,過程中配位基並未發生任何變化,參見圖一的例子。
c1
一般而言,如果反應中電子轉移的速率比配位基取代來得快,反應會循外圈電子轉移的機制,通常發生在反應物較安定、配位基不活潑或是缺乏適合橋鍵配位基的情況下。{kind=link}
圖一 Fe(CN)64- 與Ir(Cl)62-的氧化還原反應-外圈電子轉移
http://www.chem.ox.ac.uk/icl/dermot/mechanism1/lecture3/default.html
http://www.chem.ox.ac.uk/icl/dermot/mechanism1/lecture3/default.html
光變色性物質(Photochromic Materials)
光變色性物質(Photochromic Materials)
臺北市立永春高級中學化學科蔡曉信老師
光變色性(photochromism)是某些種類的化學物質受到電磁波(光線)的照射而變化成另一種物質,其反應為可逆反應且兩物質的顏色不同。這個現象最早是在1880年代由Markwald 研究2,3,4,4-tetrachloronaphthalen-1(4H)-one的顏色改變所發現,當時他稱此現象為” phototropy”。直到1950年代Hirshberg才正式提出光變色性(photochromism)這個專有名詞。光變色性的現象可以在有機、無機物質甚至在生物系統中發生,如視網膜(retinal)產生視覺的過程。光變色性是一種可逆的化學反應,這是一個很重要的判斷基準。在光作用下發生的不可逆反應,也能導致顏色的變化,但這只能當成一般的光化學反應,而不屬於光變色性反應。光變色性物質的種類(classes of photochromic materials)有:大都集中在二苯基乙烯(diarylethenes )、光變色性醌(photochromic quinones)、螺吡喃(spiropyrans)、螺1,4-氧氮六圜(spirooxazines)、偶氮類(azobenzenes)以及相關的雜環化合物(pericyclic compound)等。以下是常見的五種光變色性物質:
1.螺吡喃(spiropyrans)與螺1,4-氧氮六圜(spirooxazines)
b1
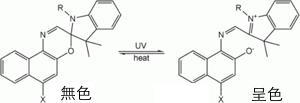
最早為科學家所研究的光變色性物質為螺吡喃,與其密切相關的化學物質為螺1,4-氧氮六圜。例如:螺環化合物1,4-氧氮六圜為無色的染料,1,4-氧氮六圜的共軛系統與其它部分的芳香環間以sp3混成軌域的C原子聯結。當此分子被紫外光照射時其C-O鍵會斷裂使其結構中的六元環開環,因此其原本sp3混成軌域的C原子就轉變成sp2混成軌域而成為平面結構。這樣的轉變使其結構中的共軛雙鍵數增加,因此可吸收可見光而有顏色的呈現。反之當外在的紫外線消失時,其分子又轉變成較低能量的基態,此時芳香環間C原子的sp3混成軌域再度形成,顏色又變回無色。
臺北市立永春高級中學化學科蔡曉信老師
光變色性(photochromism)是某些種類的化學物質受到電磁波(光線)的照射而變化成另一種物質,其反應為可逆反應且兩物質的顏色不同。這個現象最早是在1880年代由Markwald 研究2,3,4,4-tetrachloronaphthalen-1(4H)-one的顏色改變所發現,當時他稱此現象為” phototropy”。直到1950年代Hirshberg才正式提出光變色性(photochromism)這個專有名詞。光變色性的現象可以在有機、無機物質甚至在生物系統中發生,如視網膜(retinal)產生視覺的過程。光變色性是一種可逆的化學反應,這是一個很重要的判斷基準。在光作用下發生的不可逆反應,也能導致顏色的變化,但這只能當成一般的光化學反應,而不屬於光變色性反應。光變色性物質的種類(classes of photochromic materials)有:大都集中在二苯基乙烯(diarylethenes )、光變色性醌(photochromic quinones)、螺吡喃(spiropyrans)、螺1,4-氧氮六圜(spirooxazines)、偶氮類(azobenzenes)以及相關的雜環化合物(pericyclic compound)等。以下是常見的五種光變色性物質:
1.螺吡喃(spiropyrans)與螺1,4-氧氮六圜(spirooxazines)
b1
{kind=link}
最早為科學家所研究的光變色性物質為螺吡喃,與其密切相關的化學物質為螺1,4-氧氮六圜。例如:螺環化合物1,4-氧氮六圜為無色的染料,1,4-氧氮六圜的共軛系統與其它部分的芳香環間以sp3混成軌域的C原子聯結。當此分子被紫外光照射時其C-O鍵會斷裂使其結構中的六元環開環,因此其原本sp3混成軌域的C原子就轉變成sp2混成軌域而成為平面結構。這樣的轉變使其結構中的共軛雙鍵數增加,因此可吸收可見光而有顏色的呈現。反之當外在的紫外線消失時,其分子又轉變成較低能量的基態,此時芳香環間C原子的sp3混成軌域再度形成,顏色又變回無色。
No comments:
Post a Comment