1995年,埃里克·本茨格得出结论,他认为近场光学显微镜已经没有多少进行进一步改进的空间。除此之外,他也感到自己并不适合学术界,因此决定结束自己的学术生涯。但他对自己接下来要去哪里感到迷茫。他从贝尔实验室离职了,但有关Abbe衍射极限的问题仍然萦绕在他的脑海之中。在一个冬日的散步期间,他突然产生了一个新的念头:是否有可能利用不同分子的不同特性来避开Abbe衍射极限的问题,比如利用那些能够产生不同颜色荧光的分子?
受到默尔纳和其他科学家研究的启发,埃里克·本茨格此前已经利用近场光学显微镜观察到了单分子的荧光现象。现在他开始思考这样一个问题:如果不同的分子发出不同颜色的荧光,比如红色,黄色和绿色,那么利用常规显微镜是否也有可能达成这样高的分辨率?他的具体想法是让显微镜每次只记录一种颜色。如果所有发出同一种颜色的分子都散布开来,它们之间的间距都超过Abbe衍射极限所限定的0.2微米,那么这些单个分子的所谓位置便能够被非常精确的确定下来。下一步,当不同颜色下记录的图像被叠加在一起,那么此时得到的图像分辨率将会大大超越Abbe衍射极限所限定的水平,而红色,黄色和绿色的分子即便它们各自之间的距离仅有几个纳米,此时将仍然可以被分辨出来。通过这种方法,Abbe的衍射极限问题就可以被绕开了。然而还存在一些实际问题,比如他找不到具有足够可区分光学特性的分子。
1995年,埃里克·本茨格在《光学通报》杂志上报告了自己的理论设想,并在随后离开了学术界并到他父亲的公司工作去了
详解2014诺贝尔化学奖:超越光学显微成像极限
对光学显微镜的分辨率限制做出了界定,认为大约是光波长的一半,即约为0.2微米。这意味着科学家们可以辨别完整细胞,以及其中一些被称为细胞器的组成部分。然而,他们却无法分辨一个正常大小的病毒或者单个蛋白质。") 图一: 在19世纪末,恩斯特•阿贝(Ernst Abbe)对光学显微镜的分辨率限制做出了界定,认为大约是光波长的一半,即约为0.2微米。这意味着科学家们可以辨别完整细胞,以及其中一些被称为细胞器的组成部分。然而,他们却无法分辨一个正常大小的病毒或者单个蛋白质。
图一: 在19世纪末,恩斯特•阿贝(Ernst Abbe)对光学显微镜的分辨率限制做出了界定,认为大约是光波长的一半,即约为0.2微米。这意味着科学家们可以辨别完整细胞,以及其中一些被称为细胞器的组成部分。然而,他们却无法分辨一个正常大小的病毒或者单个蛋白质。 图2:在常规光学显微镜中,可以区分线粒体的轮廓,但其分辨率却无法超越0.2微米。
图2:在常规光学显微镜中,可以区分线粒体的轮廓,但其分辨率却无法超越0.2微米。使用STED显微镜拍摄而成的图像。左边为使用传统显微镜拍摄的大肠杆菌,右边是使用STED显微镜拍摄的同样的大肠杆菌。STED图像的分辨率是前者的3倍") 图3:第一张由斯特凡•W•黑尔(Stefan W. Hell)使用STED显微镜拍摄而成的图像。左边为使用传统显微镜拍摄的大肠杆菌,右边是使用STED显微镜拍摄的同样的大肠杆菌。STED图像的分辨率是前者的3倍
图3:第一张由斯特凡•W•黑尔(Stefan W. Hell)使用STED显微镜拍摄而成的图像。左边为使用传统显微镜拍摄的大肠杆菌,右边是使用STED显微镜拍摄的同样的大肠杆菌。STED图像的分辨率是前者的3倍 图4:单分子显微镜原理
图4:单分子显微镜原理,是埃里克•白兹格(Eric Betzig)首次使用单分子显微镜拍摄的图片之一。从中选取0.2微米的阿贝衍射极限大小显示在右边。左边为用传统显微镜拍摄的图片,可以看出图片分辨率提高了很多倍。") 图5:中间图像为溶酶体膜(lysosome membranes),是埃里克•白兹格(Eric Betzig)首次使用单分子显微镜拍摄的图片之一。从中选取0.2微米的阿贝衍射极限大小显示在右边。左边为用传统显微镜拍摄的图片,可以看出图片分辨率提高了很多倍。
图5:中间图像为溶酶体膜(lysosome membranes),是埃里克•白兹格(Eric Betzig)首次使用单分子显微镜拍摄的图片之一。从中选取0.2微米的阿贝衍射极限大小显示在右边。左边为用传统显微镜拍摄的图片,可以看出图片分辨率提高了很多倍。光学显微成像技术向纳米尺度的迈进
血红细胞,细菌,酵母菌以及游动的精子。当17世纪的科学家们第一次在光学显微镜下看到这些活生生的生物现象时,一个崭新的世界在他们的眼前打开了。这就是光学显微成像技术的诞生。自那以后,光学显微镜已经成为生物学研究领域最重要的工具之一。其他显微成像技术,如电子显微镜,都需要进行样品的制备,而这样的制备过程会杀死细胞。
借助分子发光技术超越物理极限
然而,长期以来,光学显微成像技术的发展却一直受制于一个物理极限值的约束。1873年,显微技术专家Ernst Abbe提出了传统显微成像技术的物理极限值:这种技术的分辨率将永远不能超过0.2微米。这一预言导致在20世纪的绝大多数时间里,科学家们都相信光学显微成像技术将永远无法让他们突破到更细微的尺度上(Fig 1)。一些细胞内部的细胞器,如为细胞活动提供能量的线粒体,它们的轮廓是可以看到的。但要想进一步观察更小的对象,如细胞内部单个分子之间的相互作用则是根本不可能做到的。这就有点像是观察一座城市,你可以看到城市里林立的高楼,但却无法看清其中生活的居民们进进出出的日常生活。为了了解细胞的日常运作,科学家们需要对单个分子的活动进行追踪。
然而Abbe提出的这一物理极限由于今年的诺贝尔化学奖获奖人的工作被突破了。从理论上说,现在再也没有任何障碍,阻止科学家们对更小尺度上的物体进行观察了。于是,显微成像变成了纳米显微成像。
在突破Abbe极限的方案中,有两种各自独立发展出来的技术方法。而整个故事还得从1993年位于芬兰西南部的一间学生宿舍里讲起。有一天,当史蒂芬·赫尔在翻阅一本量子光学书时,他想到了一个绝妙的主意。
直面Abbe极限的年轻挑战者
1990年在海德堡大学获得博士学位之后,史蒂芬·赫尔一直在设想超越一个多世纪前提出的Abbe极限的方法。想要挑战一种现存的主流观点是令人兴奋的。但当时德国的几乎所有顶尖科学家都对他的想法持怀疑态度,于是赫尔转而向遥远的北方寻求支持。芬兰图尔库大学一名主攻荧光显微成像技术的教授给了他在自己研究组里的一个职位。赫尔坚信一定有着可以突破Abbe极限的方法。而当他在一本量子光学书中读到有关受激发射的内容时,一种全新的想法在他的脑海中逐渐成型。2009年时他曾经这样评价当时自己的感受:“当时,那个想法吸引了我。我终于有了明确的想要去追求的方向。”
解决方案:纳米尺度的闪光样品扫描
在芬兰图尔库大学,赫尔专攻所谓荧光显微成像学,这是一种技术方法,科学家们借助荧光分子对细胞的局部进行成像。比如说他们可以利用荧光抗体结合的方法来观察分子DNA。他们利用短暂的闪光激发该抗体,让它们在短时间内发光。如果抗体与DNA结合了,那么它就会从细胞的核心部位发光,因为那里正是细胞核内存储DNA的位置。通过这种方法,科学家们可以判断某一特定分子的所在位置。但这样也仅仅是能让科学家们确定大团分子,如缠绕纠缠的DNA分子的所在位置。这样做的分辨率太低,难以区分出特定的DNA链。你可以想象一下看到缠绕的纱线,而看不清单根纱线的场景。
而当史蒂芬·赫尔读到有关受激发射的内容时,他意识到应当有可能制成一种装置,其使用纳米闪光灯扫过样品,每次一纳米。利用受激发射原理,科学家们可以冷却荧光分子。它们将一束激光束对准一个分子,后者立即失去能量并变得暗淡。在1994年,史蒂芬·赫尔发表了一篇文章陈述了自己的这一想法。在这一他设想中的技术方案,也就是所谓“受激发射减损技术”(STED)中计划采用闪光来激发所有的荧光分子,随后利用另外一次闪光让所有分子荧光熄灭——那些位于中部位置上纳米尺度空间内的除外(图 2)。当进行记录时则只记录下这一部分。让这一光束扫过整个样品表面,并连续记录光强信息,就有可能得到一张整体图像。每次允许发出荧光的空间区域越小,最后得到的图像分辨率便越高。于是,从原理上说,对于光学显微成像的极限再也不复存在了。
在德国研制首台纳米闪光装置
然而史蒂芬·赫尔的理论文章并没有立即在学术界引起关注,但却足以让史蒂芬·赫尔在德国马克斯普朗克生物物理化学研究所获得一个职位,在接下来的数年里,他将自己的设想逐渐变成现实:他开发出了STED显微镜。2000年,他证明了自己的技术方法在实际工作中是可行的。当时他对大肠杆菌进行了摄像,其分辨率是此前任何光学显微镜都从来未能达到过的(图 3)。
STED显微镜通过多次小区域上采集光线并最终形成整体成像图。与之相反,此次获奖的第二种技术方案,即所谓“单分子显微成像技术”,则涉及多幅整体图像的叠加。埃里克·本茨格和威廉·默尔纳各自独立的奠定了这项技术的基础。这一技术的最初故事是从默尔纳首次成功探测到单个微小的荧光分子开始的。
默尔纳——首次探测到单个荧光分子
在大多数化学方法中,比如测量荧光的吸收,科学家们一般都是同时对数以百万计的分子同时进行观察。这类实验得到的结果一般代表的是典型或平均分子的情况。科学家们只能接受这样的结果,因为这是无法改变的。但长期以来他们都一直梦想着有朝一日能够对单一分子进行直接的测量,因为更加详尽和丰富的认识或许能够带来对一些现象,比如疾病发展过程更深刻的理解。
于是,在1989年,当默尔纳成为世界上首位成功测量单个荧光分子光吸收的科学家时,那是一项伟大的成就。当时他在IBM位于加州的研发中心工作。这项实验开启了通往新的未来的大门,并启发大量化学家将他们的注意力转向单分子研究,其中一位便是埃里克·本茨格。
8年之后,默尔纳朝着单分子显微成像技术又向前迈进了一步,他在此前曾经被授予诺贝尔奖的绿色荧光蛋白技术(GFP)的基础上进行了发展。
带着开关的小灯泡
1997年,默尔纳来到加州大学圣迭戈分校,后来被授予诺贝尔奖获,GFP技术的发明人钱永佑教授也在这里任职。钱教授从水母体内分离出发绿色荧光的蛋白,其重要意义在于它能够让活体生物体内细胞的其他蛋白质同样变得可见。运用基因技术,科学家们将这些绿色荧光蛋白与其他类型的蛋白进行耦合。这样,利用绿色荧光作为标记,科学家们便能知道那些被标记的蛋白质在生物体内所处的位置。
默尔纳注意到GFP荧光分子的一种,其荧光可以被随意的开启或关闭。当他用波长488纳米的光线激发这一蛋白质时,它开始发出荧光,但一会之后它就熄灭了。此后不管他再使用多少光线去照射它,这个蛋白质的荧光都已经死了。然而,此后他发现,如果使用波长为405纳米的光线去照射它,那么这个蛋白质又能再次复活并发出荧光。当该蛋白质被再次激活,它会再次发出波长为488纳米的荧光。
默尔纳将这些可以被激发的蛋白质融入一种溶胶,使其均匀散布其中,这样其单个分子之间的距离就能大于当年Abbe衍射极限所限定的0.2微米的长度。由于这些分子被分散了开来,一台常规的光学显微镜便可以区分来自单个分子发出的荧光——它们就像是带着开关的微小灯泡。有关这一实验的结果被发表在1997年的一期《自然》杂志上。
通过这一发现,默尔纳显示了通过光学手段操控单个分子荧光的可能性。这一结果解决了埃里克·本茨格两年来一直困扰着他的问题。
厌倦了学术界,但仍对Abbe衍射极限感到着迷
与史蒂芬·赫尔一样,埃里克·本茨格对于突破Abbe设下的衍射极限非常着迷。1990年代初,本茨格正在美国新泽西州贝尔实验室开展一种名叫“近场光学显微镜”的研究工作。在近场光学显微镜中,光线是从极为贴近样品表面,距离仅有几个纳米的薄片上发出的。这种显微镜可以突破Abbe的衍射极限,但这一方法也有着难以克服的缺陷。比如说,其产生的光线作用距离极短,因此难以呈现细胞表面以下的深处结构。
1995年,埃里克·本茨格得出结论,他认为近场光学显微镜已经没有多少进行进一步改进的空间。除此之外,他也感到自己并不适合学术界,因此决定结束自己的学术生涯。但他对自己接下来要去哪里感到迷茫。他从贝尔实验室离职了,但有关Abbe衍射极限的问题仍然萦绕在他的脑海之中。在一个冬日的散步期间,他突然产生了一个新的念头:是否有可能利用不同分子的不同特性来避开Abbe衍射极限的问题,比如利用那些能够产生不同颜色荧光的分子?
受到默尔纳和其他科学家研究的启发,埃里克·本茨格此前已经利用近场光学显微镜观察到了单分子的荧光现象。现在他开始思考这样一个问题:如果不同的分子发出不同颜色的荧光,比如红色,黄色和绿色,那么利用常规显微镜是否也有可能达成这样高的分辨率?他的具体想法是让显微镜每次只记录一种颜色。如果所有发出同一种颜色的分子都散布开来,它们之间的间距都超过Abbe衍射极限所限定的0.2微米,那么这些单个分子的所谓位置便能够被非常精确的确定下来。下一步,当不同颜色下记录的图像被叠加在一起,那么此时得到的图像分辨率将会大大超越Abbe衍射极限所限定的水平,而红色,黄色和绿色的分子即便它们各自之间的距离仅有几个纳米,此时将仍然可以被分辨出来。通过这种方法,Abbe的衍射极限问题就可以被绕开了。然而还存在一些实际问题,比如他找不到具有足够可区分光学特性的分子。
1995年,埃里克·本茨格在《光学通报》杂志上报告了自己的理论设想,并在随后离开了学术界并到他父亲的公司工作去了。
重返学术界
有很多年时间,埃里克·本茨格与学术界完全脱离了。但有一天,渴望科学的种子再次在他身体里萌发了,于是他的目光再次回到了科学领域,这一次他注意到了关于绿色荧光分子的消息。他很快意识到这种可以让活体细胞内其他蛋白质发光的荧光蛋白质或许可以被用来绕开Abbe衍射极限。
真正的突破出现在2005年,这一年他无意间发现了一种可以随意开启或关闭其荧光的蛋白质,跟默尔纳在1997年在但分子层面上所观察到的情况很像。本茨格意识到这正是实施10多年前他头脑中那个想法所缺少的工具。荧光分子并不一定需要具有不同的颜色,它们只要能在不同的时间发出荧光就可以了。
通过图像叠加方法突破Abbe衍射极限
短短一年之后,埃里克·本茨格与其他研究激发荧光蛋白的科学家们一起,证明了他的技术方案在实践中是可行的。在许多不同的实验中,有一项实验是将该蛋白质与溶酶体外膜结合,这里是细胞的回收站。在光信号刺激下,蛋白质发出荧光,但由于光线非常弱,只有一部分的蛋白质发光。由于数量少,几乎每个分子之间的距离都要超过Abbe衍射极限所限定的0.2微米长度。于是,在显微镜下,每一个发光分子的位置都可以被非常精确的记录下来。过了一会,等到这些分子的荧光逐渐熄灭之后,研究组再激活另外一组蛋白质分子,让它们发出荧光。同样的,只有一部分分子会发光,同样的,记录下每一次发光分子的图像。这一过程被一再重复。
探索仍在继续
这两种分别由埃里克·本茨格,史蒂芬·赫尔以及威廉·默尔纳发展出来的技术方法自那以后已经导致多项纳米尺度成像科技的诞生,目前已经在世界各地得到广泛应用。目前这三位获奖人仍然活跃在这里研究领域的第一线,与越来越多投身这一研究领域的科学家们一道继续开展工作。当他们将强大的纳米成像设备对准组成生命的最微小组件,他们帮助人们获得了大量最新的知识。史蒂芬·赫尔目前正致力于对神经细胞的精密探查,以便更好加深对大脑突触的理解;威廉·默尔纳正在开展与亨廷顿综合征有关蛋白质的研究,而埃里克·本茨格正在努力追踪胚胎内部细胞分裂的过程。这些都还只是他们正在从事的大量工作中的几个案例。
但有一件事是可以肯定的,那就是2014年度的诺贝尔化学奖获得者们已经为我们奠定了坚实的基础,去追寻人类最为重要的知识。(晨风)
如何通俗地理解 2014 年诺贝尔化学奖「超分辨荧光显微技术」的技术原理及其带来的改变?
「超分辨荧光显微技术」指的是哪些呢?与普通的荧光显微技术相比,实现了哪些技术突破?以及具体应用于哪些工作场景?为该工作的开展带来了怎么样的改变?
此问主要从技术层面讨论,如不过瘾还请关注系列问题:
2014 年诺贝尔化学奖「超分辨荧光显微技术」的「化学」含量有多少?
如何评价2014年诺贝尔化学奖?
此问主要从技术层面讨论,如不过瘾还请关注系列问题:
2014 年诺贝尔化学奖「超分辨荧光显微技术」的「化学」含量有多少?
如何评价2014年诺贝尔化学奖?
按票数排序按时间排序
7 个回答
私货已经在另一个答案(如何评价2014年诺贝尔化学奖?)里面吐槽过了,来认真写个答案吧。第一次写同时面向非专业和专业人士的科普长文,希望没看明白的同学留言指出,我尽量修改,谢谢!
------------------------------------------------
2014 年的诺贝尔理综奖颁发给了“超分辨荧光显微技术”。也许接下来的几天,媒体会关注 Stefan Hell、Eric Betzig 二人的传奇经历,或者另一名华人女科学家与该奖项失之交臂的遗憾。但是八卦之外,这项成果背后的科学本身也非常有意思。
这里面有三个关键词:“超分辨”、“荧光”和“显微技术”,我希望能够解释清楚以下几个问题,尤其是后两个问题:
1. 为什么需要(光学)显微技术?
2. 为什么光学显微镜的分辨率存在理论极限?
3. 用怎样的方法可以突破这个理论极限以达到“超分辨”?为什么这个理论极限可以被突破?
4. 为什么非得是荧光显微技术,而非普通的明场(透射光)显微技术?
1. 采样定理与显微镜
我们用肉眼观察或者用相机拍摄一个物体时,物体上的每一个细微的点都会在眼睛的视网膜或是相机的感光芯片上成像。那么我们为什么不能看到细菌等微小的东西,为什么不能把照片无限放大以看清远处树木上面的每一片叶子呢?
这个问题的答案比较简单:因为组成视网膜的每一个感光细胞(视杆细胞和视锥细胞)、相机芯片上的每一个感光元件(CCD、CMOS等)都是有大小的。比如视网膜中央凹区域的视锥细胞直径平均约为 5 微米。而由于奈奎斯特-香农采样定理的限制,视网膜上能分清的两个相邻像点的距离是视锥细胞直径的两倍,即 10 微米。再结合眼球的构造,大致可以推断出,在距离眼睛 25 厘米的位置,我们能分辨物体上相距为 80 微米的两个点,换算成点阵密度就是大约 320 ppi,这也是苹果所谓“视网膜屏”分辨率的来历。
如果要观察小于 80 微米的物体,比如细菌,就需要先将物体放大,再用眼睛或者相机观察。现代光学显微镜的构造其实非常简单,样品放置在物镜的焦点处,从样品上发射或散射的光经过物镜变成平行(准直)光,再经过一个结像透镜,然后会聚到相机的感光芯片上成像。
按照前面的方法来推算,要区分物体上相距为 200 纳米的两个点,如果使用科研级相机,比如最近火起来的 sCMOS 相机(每个感光像素尺寸为 6.5 微米),只需要使用放大倍率为 65 倍的物镜就足够了。
那么是否可以通过提高物镜的放大倍率来观察低于 200 纳米的物体,比如细胞里面微管呢?
答案是不可以。
2. 光学衍射极限
由于光是一种电磁波,具有衍射和干涉的特性。
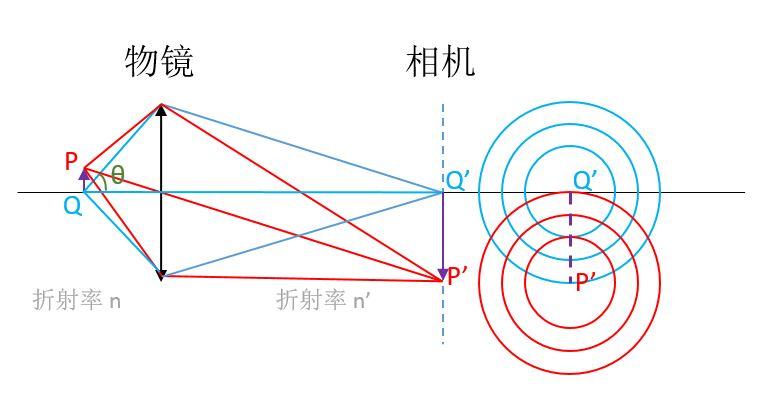 图 1. 光学显微镜简化示意图
图 1. 光学显微镜简化示意图
如上面的简图所示,紫色箭头表示的物体 PQ 经过物镜等之后在相机上成像为P'Q'。由于光的衍射,物体上的点如 P、Q,在相机上并不是单独的点,而是一个个有一定大小的斑,被称为夫琅禾费衍射斑(或称艾里斑),如右侧的同心圆所示。那么,当 P'、Q' 相距太近的时候,两个斑会叠加导致难以分辨。这就要求物体上的 P、Q 要相距一定的距离。
1873 年,德国物理学家、卡尔蔡司公司的恩斯特·阿贝(Ernst Abbe)首次推算出衍射导致的分辨率极限。根据瑞利判据——“当一个像斑的中心落到另一个像斑的边缘时,就算这两个像刚好能被分辨”,显微镜能分辨的物体上两点 P、Q 的最小距离 h 为:

这个公式就是光学显微镜的分辨率公式,或称为光学衍射极限。(注意此处的分辨率与通常说的显示器分辨率含义不同)
其中, 为光的波长,n 为物方的折射率,
为光的波长,n 为物方的折射率, 为物体与物镜边缘连线和光轴的夹角。如上图所示。为方便起见,其中的
为物体与物镜边缘连线和光轴的夹角。如上图所示。为方便起见,其中的  通常称为数值孔径,简写为 NA。
通常称为数值孔径,简写为 NA。
摄影领域常用 f/# 或称光圈值来描述镜头,光圈值与数值孔径可以相互换算。对于一枚光圈为 2.0 的镜头来说,数值孔径为 0.25,其分辨能力约 1.2 微米。
目前常用的高倍物镜 NA 最大为1.49,可以算出,对于可见光,比如波长 500 纳米的绿光,显微镜的分辨率约为 200 纳米。所以,即使再提高物镜的放大倍率,也不能提高显微镜的分辨率。
而 200 纳米这个数值也就通常被称作衍射极限。
2.5. 光学衍射极限的动态范围
从上面的公式可以看到,光学衍射极限其实并不是一个具体的数值,如果改变上述公式中的参数,是可以有效提高分辨率的。
(1) 光的波长
可见光波长范围是400~760 nm,如果使用更短波长的光,比如紫外线,理论上可以提高分辨率。但是紫外线能量高,易损伤样品,而且透射能力低,很难透过物镜。人们想到了使用高能电子束代替光束,比如 200 keV 的电子对应的的布罗意波长为 0.0025 纳米 (2.5 * 米)。虽然 NA 相对较小(约为10°),依然可以达到0.1 纳米的理论分辨率。这就是电子显微镜的基本原理。
米)。虽然 NA 相对较小(约为10°),依然可以达到0.1 纳米的理论分辨率。这就是电子显微镜的基本原理。
严格的说,电镜的分辨率依然限制在光学衍射极限的范围内。只不过这里的“光学”是“电子光学”。
(2) 物方折射率 n
空气折射率为 1,水的折射率 1.33,玻璃折射率 1.58。目前主要的物镜都是玻璃材质,并在物镜与样品之间用与玻璃折射率一致的油来浸润,以提高分辨率。
2012 年 Olympus 发布了一款 NA 高达 1.7 的物镜,光学部分使用蓝宝石(折射率约 1.76)制作,并搭配高折射率的镜油(目测成分应该是二碘甲烷 Methylene iodide)。
也许在未来能发明比玻璃更好的材料,折射率更高、易于制作透镜、并且能找到高折射率的油,这样就能进一步提高分辨率。
比如用钻石(折射率大约2.42)打造一枚土豪物镜,并找到同样折射率的透明液体,分辨率可以提高到 1.5 倍。当然,由于成本及工艺因素,目前尚不现实。
(3) 孔径角
看起来 的最大值是 90°,但是如果在样品的上下两面都放置物镜,相机同时收集这两个物镜中的光呢?
这种方法就是 Stephen Hell 在 1991 年发明的 4 Pi 显微技术。此时衍射极限的公式稍有变化,分辨率能提高一倍。
简单理解,限制分辨率的一个原因是从物体上向四面八方发射的光线在经过透镜时,由于透镜大小(孔径)的限制,所以很多光线没有经过物体,其承载的物体的“信息”丢失了。而 4 Pi 其实是通过两个物镜收集了更多的光及“信息”。每个高 NA 的物镜离物体都非常近,所以几乎能收集到各个角度的光,这也是 4 Pi 这个名称的来历——一个以物体为球心的球体其立体角的大小就是 。
。
另一种“结构照明显微技术(SIM)”很聪明地通过类似摩尔纹的原理,获取到更多“信息”,也可以将分辨率提高一倍。解释原理需要用到傅里叶光学,在此就不详述了。它由已故的 Mats Gustafsson 教授于 2000 年发明。
那么问题来了,是否还有办法“真正”突破衍射极限,使之并不受限于这个公式呢?
3. 突破光学衍射极限
前面的公式推导过程中,有两个隐含条件。
第一个隐含条件是需要用到夫琅禾费衍射,而它也是有条件的,它要求物体与像平面之间的距离远远大于光的波长。这个条件称为远场条件。如果离物体足够近,衍射极限便不遵循上述公式。研究波长以内光传播特性的领域叫近场光学。
Eric Betzig 在 1986 年发明的近场扫描光学显微技术(NSOM)就是使用距离物体远小于波长的光学探针来扫描物体,以达到超分辨的显微图像。
4. 单分子荧光与分辨率革命(revolution of resolution)
另一个条件则非常之隐蔽,但却十分朴素,以至于自阿贝推算出衍射极限公式之后的 100 多年间,没有人能够发现或重视。
Stefan Hell 与 Eric Betzig 的贡献就在于解锁了这个隐藏关卡,在固定的波长与 NA 之下,将显微镜的理论分辨能力提高到无限大,带来了 1990 年代以来的分辨率革命。而这次革命非常依赖于 William Moerner 推动的单分子荧光成像等研究,也是为什么本次诺奖会颁发给几乎没做过超分辨显微技术的 William Moerner、颁发给理综奖的原因。
这里要简单插播一下荧光成像的原理:荧光分子(如获得 2008 年的诺贝尔理综奖的绿色荧光蛋白 GFP)在吸收一个高能量的短波长(如蓝光,称为激发光)跃迁到高能态之后,很快会发射一个低能量的长波长(如绿光,称为发射光)光子回到基态能级。
所以我们可以用蓝光照射样品上标记的 GFP,而接收其发射的绿光,来对物体进行成像。这样的一个好处就是图像对比度会非常高,只有带有 GFP 的部位会被成像,其它部位都是暗的。
而每个荧光分子的发光是完全独立的。
回到前面说的第二个隐藏条件,说穿了其实非常简单。推导时假定了图 1 中的物体上的两点 P、Q 同时发光,所以二者的衍射斑才会叠加在一起导致难以区分。但如果 P、Q 是在不同的时间分别发光,那么可以(通过点扩散函数拟合或者去卷积的方法)精确地定位到每个衍射斑的中心点位置 P'、Q'。那么也就不存在这样的衍射极限了,理论上即使 P、Q 相距再近都可以被分别定位而加以区分。
更进一步,广义地来说,只要是 P、Q 两点处在可以探测到的不同“状态”或是不同特征,那么就可以超越衍射极限。分辨率并不仅仅是关于波的,而且是关于“状态”的!
意识到了这一点,很多种真正突破衍射极限的显微技术就被解锁了。比如:
(1) 区分荧光分子的不同构象/能态
荧光分子处在不同构象或能态时,可以发射不同波长的光子,或者不发光。
受激发射耗损显微术(STED):激发光周围套上一圈耗损光,使中心区域的荧光分子处于激发态发光,周围的分子处于耗损态不发光。这样可以使图 1 中的同心圆表示的衍射斑变小,以此来区分两个相距在衍射极限以内的分子。这个方法由 Stefan Hell 在 1994 年发明。
随机光学重构显微术(STORM)、光活化定位显微术(PALM)、荧光活化定位显微术(fPALM):它们的共同特征是使样品中每次仅有少量随机、离散的单个荧光分子发光,通过拟合,找出每个荧光分子中心点的位置。重复拍摄多张图片之后,就可以把所有荧光分子的中心点位置叠加起来形成整幅图像。这些技术由庄小威、Eric Betzig 等人在 2006 分别独立发明。

图 2. 庄小威发明的 STORM 超分辨光学成像效果图。a, b 为传统光学显微镜拍摄,c, d 为相同区域经 STORM 技术拍摄并重构。b, d 为 a, c 的局部放大。图中的结构是细胞内的支架——微管蛋白。图片来自 Nikon MicroscopyU 。
RESOLFT:由于 STED 中,耗损光需要很强的亮度,激光器造价昂贵,而且对样品损伤较大,如果借鉴 STORM/PALM/fPALM 的原理,不需要使周围分子处于“耗损态”,而是采用类似于 STORM 等的特殊荧光分子,用较弱的光强使其处于其它能态,也可以实现超分辨。这也是 Stefan Hell 发明的。
(2) 荧光分子闪烁的不同周期
SOFI:有些荧光分子在持续的激发光照射下,并不是持续发光,而是会闪烁。由于每个荧光分子随机闪烁的时间不一样,可以通过连续拍摄一段录像,分析比较图像中每个像素点记录到的荧光强度在这段时间内的闪烁情况,来区分不同的荧光分子,实现超分辨。2009 年发明。
(3) 荧光分子的极性/朝向不同
SPoD/ExPAN:荧光分子都是偶极子,可以理解成有着不同的朝向。当用不同朝向的偏振光激发时,荧光分子的亮度也会随之不同。可以周期性地改变偏振光朝向,连续拍摄一段录像,分析比较图像中每个像素点荧光强度的周期变化情况,来区分不同荧光分子,实现超分辨。2014 年发明。
……
这些不同的方法,也都有着各自分辨率的计算公式。STED/RESOLFT 的理论分辨率与激发光、耗损光强度有关;STORM/PALM 与单个荧光分子发射的光子数有光;SOFI/SPoD 与相机的像素尺寸有关(采样定理决定)。在固定的波长、NA 下,理论上的分辨率依然可以达到任意高。
如果还能想到荧光分子的更多特性并加以利用,未来还会有越来越多的超分辨显微术被发明。
所以可以看到,William Moerner 开创的单分子荧光成像在超分辨成像中的作用。Stefan Hell、Eric Betzig 使用物理方法利用了荧光分子的化学特性而发明的超分辨显微成像,目前在生物学领域已经得到了广泛应用,因此,给三人颁发一个诺贝尔理综奖也是名至实归的。
------------------------
有兴趣了解超分辨实现方法细节的同学,推荐 @Hydro Ding 的这篇回答,比我写的清楚:光学显微镜受光线的波长限制只能够放大两千倍左右的极限,有办法解决么?或者这就是自然定律的一种界限表现? 显示全部
------------------------------------------------
2014 年的诺贝尔理综奖颁发给了“超分辨荧光显微技术”。也许接下来的几天,媒体会关注 Stefan Hell、Eric Betzig 二人的传奇经历,或者另一名华人女科学家与该奖项失之交臂的遗憾。但是八卦之外,这项成果背后的科学本身也非常有意思。
这里面有三个关键词:“超分辨”、“荧光”和“显微技术”,我希望能够解释清楚以下几个问题,尤其是后两个问题:
1. 为什么需要(光学)显微技术?
2. 为什么光学显微镜的分辨率存在理论极限?
3. 用怎样的方法可以突破这个理论极限以达到“超分辨”?为什么这个理论极限可以被突破?
4. 为什么非得是荧光显微技术,而非普通的明场(透射光)显微技术?
1. 采样定理与显微镜
我们用肉眼观察或者用相机拍摄一个物体时,物体上的每一个细微的点都会在眼睛的视网膜或是相机的感光芯片上成像。那么我们为什么不能看到细菌等微小的东西,为什么不能把照片无限放大以看清远处树木上面的每一片叶子呢?
这个问题的答案比较简单:因为组成视网膜的每一个感光细胞(视杆细胞和视锥细胞)、相机芯片上的每一个感光元件(CCD、CMOS等)都是有大小的。比如视网膜中央凹区域的视锥细胞直径平均约为 5 微米。而由于奈奎斯特-香农采样定理的限制,视网膜上能分清的两个相邻像点的距离是视锥细胞直径的两倍,即 10 微米。再结合眼球的构造,大致可以推断出,在距离眼睛 25 厘米的位置,我们能分辨物体上相距为 80 微米的两个点,换算成点阵密度就是大约 320 ppi,这也是苹果所谓“视网膜屏”分辨率的来历。
如果要观察小于 80 微米的物体,比如细菌,就需要先将物体放大,再用眼睛或者相机观察。现代光学显微镜的构造其实非常简单,样品放置在物镜的焦点处,从样品上发射或散射的光经过物镜变成平行(准直)光,再经过一个结像透镜,然后会聚到相机的感光芯片上成像。
按照前面的方法来推算,要区分物体上相距为 200 纳米的两个点,如果使用科研级相机,比如最近火起来的 sCMOS 相机(每个感光像素尺寸为 6.5 微米),只需要使用放大倍率为 65 倍的物镜就足够了。
那么是否可以通过提高物镜的放大倍率来观察低于 200 纳米的物体,比如细胞里面微管呢?
答案是不可以。
2. 光学衍射极限
由于光是一种电磁波,具有衍射和干涉的特性。
图 1. 光学显微镜简化示意图如上面的简图所示,紫色箭头表示的物体 PQ 经过物镜等之后在相机上成像为P'Q'。由于光的衍射,物体上的点如 P、Q,在相机上并不是单独的点,而是一个个有一定大小的斑,被称为夫琅禾费衍射斑(或称艾里斑),如右侧的同心圆所示。那么,当 P'、Q' 相距太近的时候,两个斑会叠加导致难以分辨。这就要求物体上的 P、Q 要相距一定的距离。
1873 年,德国物理学家、卡尔蔡司公司的恩斯特·阿贝(Ernst Abbe)首次推算出衍射导致的分辨率极限。根据瑞利判据——“当一个像斑的中心落到另一个像斑的边缘时,就算这两个像刚好能被分辨”,显微镜能分辨的物体上两点 P、Q 的最小距离 h 为:
这个公式就是光学显微镜的分辨率公式,或称为光学衍射极限。(注意此处的分辨率与通常说的显示器分辨率含义不同)
其中,
摄影领域常用 f/# 或称光圈值来描述镜头,光圈值与数值孔径可以相互换算。对于一枚光圈为 2.0 的镜头来说,数值孔径为 0.25,其分辨能力约 1.2 微米。
目前常用的高倍物镜 NA 最大为1.49,可以算出,对于可见光,比如波长 500 纳米的绿光,显微镜的分辨率约为 200 纳米。所以,即使再提高物镜的放大倍率,也不能提高显微镜的分辨率。
而 200 纳米这个数值也就通常被称作衍射极限。
2.5. 光学衍射极限的动态范围
从上面的公式可以看到,光学衍射极限其实并不是一个具体的数值,如果改变上述公式中的参数,是可以有效提高分辨率的。
(1) 光的波长
可见光波长范围是400~760 nm,如果使用更短波长的光,比如紫外线,理论上可以提高分辨率。但是紫外线能量高,易损伤样品,而且透射能力低,很难透过物镜。人们想到了使用高能电子束代替光束,比如 200 keV 的电子对应的的布罗意波长为 0.0025 纳米 (2.5 *
严格的说,电镜的分辨率依然限制在光学衍射极限的范围内。只不过这里的“光学”是“电子光学”。
(2) 物方折射率 n
空气折射率为 1,水的折射率 1.33,玻璃折射率 1.58。目前主要的物镜都是玻璃材质,并在物镜与样品之间用与玻璃折射率一致的油来浸润,以提高分辨率。
2012 年 Olympus 发布了一款 NA 高达 1.7 的物镜,光学部分使用蓝宝石(折射率约 1.76)制作,并搭配高折射率的镜油(目测成分应该是二碘甲烷 Methylene iodide)。
也许在未来能发明比玻璃更好的材料,折射率更高、易于制作透镜、并且能找到高折射率的油,这样就能进一步提高分辨率。
比如用钻石(折射率大约2.42)打造一枚土豪物镜,并找到同样折射率的透明液体,分辨率可以提高到 1.5 倍。当然,由于成本及工艺因素,目前尚不现实。
(3) 孔径角
看起来
这种方法就是 Stephen Hell 在 1991 年发明的 4 Pi 显微技术。此时衍射极限的公式稍有变化,分辨率能提高一倍。
简单理解,限制分辨率的一个原因是从物体上向四面八方发射的光线在经过透镜时,由于透镜大小(孔径)的限制,所以很多光线没有经过物体,其承载的物体的“信息”丢失了。而 4 Pi 其实是通过两个物镜收集了更多的光及“信息”。每个高 NA 的物镜离物体都非常近,所以几乎能收集到各个角度的光,这也是 4 Pi 这个名称的来历——一个以物体为球心的球体其立体角的大小就是
另一种“结构照明显微技术(SIM)”很聪明地通过类似摩尔纹的原理,获取到更多“信息”,也可以将分辨率提高一倍。解释原理需要用到傅里叶光学,在此就不详述了。它由已故的 Mats Gustafsson 教授于 2000 年发明。
那么问题来了,是否还有办法“真正”突破衍射极限,使之并不受限于这个公式呢?
3. 突破光学衍射极限
前面的公式推导过程中,有两个隐含条件。
第一个隐含条件是需要用到夫琅禾费衍射,而它也是有条件的,它要求物体与像平面之间的距离远远大于光的波长。这个条件称为远场条件。如果离物体足够近,衍射极限便不遵循上述公式。研究波长以内光传播特性的领域叫近场光学。
Eric Betzig 在 1986 年发明的近场扫描光学显微技术(NSOM)就是使用距离物体远小于波长的光学探针来扫描物体,以达到超分辨的显微图像。
4. 单分子荧光与分辨率革命(revolution of resolution)
另一个条件则非常之隐蔽,但却十分朴素,以至于自阿贝推算出衍射极限公式之后的 100 多年间,没有人能够发现或重视。
Stefan Hell 与 Eric Betzig 的贡献就在于解锁了这个隐藏关卡,在固定的波长与 NA 之下,将显微镜的理论分辨能力提高到无限大,带来了 1990 年代以来的分辨率革命。而这次革命非常依赖于 William Moerner 推动的单分子荧光成像等研究,也是为什么本次诺奖会颁发给几乎没做过超分辨显微技术的 William Moerner、颁发给理综奖的原因。
这里要简单插播一下荧光成像的原理:荧光分子(如获得 2008 年的诺贝尔理综奖的绿色荧光蛋白 GFP)在吸收一个高能量的短波长(如蓝光,称为激发光)跃迁到高能态之后,很快会发射一个低能量的长波长(如绿光,称为发射光)光子回到基态能级。
所以我们可以用蓝光照射样品上标记的 GFP,而接收其发射的绿光,来对物体进行成像。这样的一个好处就是图像对比度会非常高,只有带有 GFP 的部位会被成像,其它部位都是暗的。
而每个荧光分子的发光是完全独立的。
回到前面说的第二个隐藏条件,说穿了其实非常简单。推导时假定了图 1 中的物体上的两点 P、Q 同时发光,所以二者的衍射斑才会叠加在一起导致难以区分。但如果 P、Q 是在不同的时间分别发光,那么可以(通过点扩散函数拟合或者去卷积的方法)精确地定位到每个衍射斑的中心点位置 P'、Q'。那么也就不存在这样的衍射极限了,理论上即使 P、Q 相距再近都可以被分别定位而加以区分。
更进一步,广义地来说,只要是 P、Q 两点处在可以探测到的不同“状态”或是不同特征,那么就可以超越衍射极限。分辨率并不仅仅是关于波的,而且是关于“状态”的!
"Resolution is about waves and states." -- by Stefan Hell
意识到了这一点,很多种真正突破衍射极限的显微技术就被解锁了。比如:
(1) 区分荧光分子的不同构象/能态
荧光分子处在不同构象或能态时,可以发射不同波长的光子,或者不发光。
受激发射耗损显微术(STED):激发光周围套上一圈耗损光,使中心区域的荧光分子处于激发态发光,周围的分子处于耗损态不发光。这样可以使图 1 中的同心圆表示的衍射斑变小,以此来区分两个相距在衍射极限以内的分子。这个方法由 Stefan Hell 在 1994 年发明。
随机光学重构显微术(STORM)、光活化定位显微术(PALM)、荧光活化定位显微术(fPALM):它们的共同特征是使样品中每次仅有少量随机、离散的单个荧光分子发光,通过拟合,找出每个荧光分子中心点的位置。重复拍摄多张图片之后,就可以把所有荧光分子的中心点位置叠加起来形成整幅图像。这些技术由庄小威、Eric Betzig 等人在 2006 分别独立发明。
图 2. 庄小威发明的 STORM 超分辨光学成像效果图。a, b 为传统光学显微镜拍摄,c, d 为相同区域经 STORM 技术拍摄并重构。b, d 为 a, c 的局部放大。图中的结构是细胞内的支架——微管蛋白。图片来自 Nikon MicroscopyU 。
RESOLFT:由于 STED 中,耗损光需要很强的亮度,激光器造价昂贵,而且对样品损伤较大,如果借鉴 STORM/PALM/fPALM 的原理,不需要使周围分子处于“耗损态”,而是采用类似于 STORM 等的特殊荧光分子,用较弱的光强使其处于其它能态,也可以实现超分辨。这也是 Stefan Hell 发明的。
(2) 荧光分子闪烁的不同周期
SOFI:有些荧光分子在持续的激发光照射下,并不是持续发光,而是会闪烁。由于每个荧光分子随机闪烁的时间不一样,可以通过连续拍摄一段录像,分析比较图像中每个像素点记录到的荧光强度在这段时间内的闪烁情况,来区分不同的荧光分子,实现超分辨。2009 年发明。
(3) 荧光分子的极性/朝向不同
SPoD/ExPAN:荧光分子都是偶极子,可以理解成有着不同的朝向。当用不同朝向的偏振光激发时,荧光分子的亮度也会随之不同。可以周期性地改变偏振光朝向,连续拍摄一段录像,分析比较图像中每个像素点荧光强度的周期变化情况,来区分不同荧光分子,实现超分辨。2014 年发明。
……
这些不同的方法,也都有着各自分辨率的计算公式。STED/RESOLFT 的理论分辨率与激发光、耗损光强度有关;STORM/PALM 与单个荧光分子发射的光子数有光;SOFI/SPoD 与相机的像素尺寸有关(采样定理决定)。在固定的波长、NA 下,理论上的分辨率依然可以达到任意高。
如果还能想到荧光分子的更多特性并加以利用,未来还会有越来越多的超分辨显微术被发明。
所以可以看到,William Moerner 开创的单分子荧光成像在超分辨成像中的作用。Stefan Hell、Eric Betzig 使用物理方法利用了荧光分子的化学特性而发明的超分辨显微成像,目前在生物学领域已经得到了广泛应用,因此,给三人颁发一个诺贝尔理综奖也是名至实归的。
------------------------
有兴趣了解超分辨实现方法细节的同学,推荐 @Hydro Ding 的这篇回答,比我写的清楚:光学显微镜受光线的波长限制只能够放大两千倍左右的极限,有办法解决么?或者这就是自然定律的一种界限表现? 显示全部
No comments:
Post a Comment