上次听了一个南洋理工大学教授的报告,在报告上大家就这个衍射极限作了一些深入的讨论。这个教授做的是用电磁波逆问题的思路来成像,简单来说,就是先测量一个物体周围的场,然后通过一个数据处理来确定这个物体的形状及电磁参数。
如何通俗地理解 2014 年诺贝尔化学奖「超分辨荧光显微技术」的技术原理及其带来的改变?
「超分辨荧光显微技术」指的是哪些呢?与普通的荧光显微技术相比,实现了哪些技术突破?以及具体应用于哪些工作场景?为该工作的开展带来了怎么样的改变?
此问主要从技术层面讨论,如不过瘾还请关注系列问题:
2014 年诺贝尔化学奖「超分辨荧光显微技术」的「化学」含量有多少?
如何评价2014年诺贝尔化学奖?
此问主要从技术层面讨论,如不过瘾还请关注系列问题:
2014 年诺贝尔化学奖「超分辨荧光显微技术」的「化学」含量有多少?
如何评价2014年诺贝尔化学奖?
按票数排序按时间排序
7 个回答
私货已经在另一个答案(如何评价2014年诺贝尔化学奖?)里面吐槽过了,来认真写个答案吧。第一次写同时面向非专业和专业人士的科普长文,希望没看明白的同学留言指出,我尽量修改,谢谢!
------------------------------------------------
2014 年的诺贝尔理综奖颁发给了“超分辨荧光显微技术”。也许接下来的几天,媒体会关注 Stefan Hell、Eric Betzig 二人的传奇经历,或者另一名华人女科学家与该奖项失之交臂的遗憾。但是八卦之外,这项成果背后的科学本身也非常有意思。
这里面有三个关键词:“超分辨”、“荧光”和“显微技术”,我希望能够解释清楚以下几个问题,尤其是后两个问题:
1. 为什么需要(光学)显微技术?
2. 为什么光学显微镜的分辨率存在理论极限?
3. 用怎样的方法可以突破这个理论极限以达到“超分辨”?为什么这个理论极限可以被突破?
4. 为什么非得是荧光显微技术,而非普通的明场(透射光)显微技术?
1. 采样定理与显微镜
我们用肉眼观察或者用相机拍摄一个物体时,物体上的每一个细微的点都会在眼睛的视网膜或是相机的感光芯片上成像。那么我们为什么不能看到细菌等微小的东西,为什么不能把照片无限放大以看清远处树木上面的每一片叶子呢?
这个问题的答案比较简单:因为组成视网膜的每一个感光细胞(视杆细胞和视锥细胞)、相机芯片上的每一个感光元件(CCD、CMOS等)都是有大小的。比如视网膜中央凹区域的视锥细胞直径平均约为 5 微米。而由于奈奎斯特-香农采样定理的限制,视网膜上能分清的两个相邻像点的距离是视锥细胞直径的两倍,即 10 微米。再结合眼球的构造,大致可以推断出,在距离眼睛 25 厘米的位置,我们能分辨物体上相距为 80 微米的两个点,换算成点阵密度就是大约 320 ppi,这也是苹果所谓“视网膜屏”分辨率的来历。
如果要观察小于 80 微米的物体,比如细菌,就需要先将物体放大,再用眼睛或者相机观察。现代光学显微镜的构造其实非常简单,样品放置在物镜的焦点处,从样品上发射或散射的光经过物镜变成平行(准直)光,再经过一个结像透镜,然后会聚到相机的感光芯片上成像。
按照前面的方法来推算,要区分物体上相距为 200 纳米的两个点,如果使用科研级相机,比如最近火起来的 sCMOS 相机(每个感光像素尺寸为 6.5 微米),只需要使用放大倍率为 65 倍的物镜就足够了。
那么是否可以通过提高物镜的放大倍率来观察低于 200 纳米的物体,比如细胞里面微管呢?
答案是不可以。
2. 光学衍射极限
由于光是一种电磁波,具有衍射和干涉的特性。
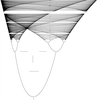
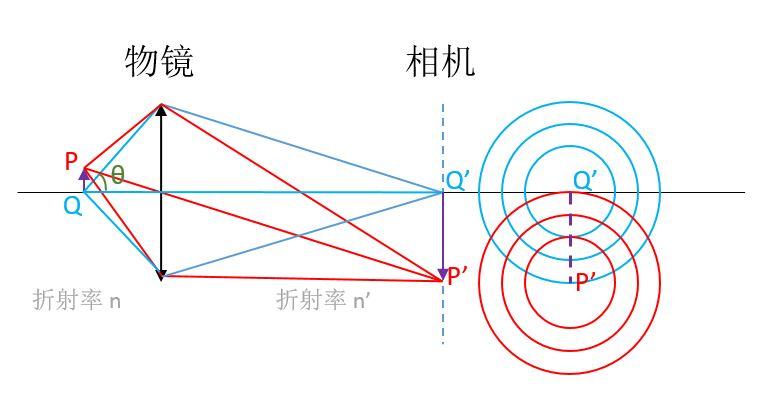 图 1. 光学显微镜简化示意图
图 1. 光学显微镜简化示意图
如上面的简图所示,紫色箭头表示的物体 PQ 经过物镜等之后在相机上成像为P'Q'。由于光的衍射,物体上的点如 P、Q,在相机上并不是单独的点,而是一个个有一定大小的斑,被称为夫琅禾费衍射斑(或称艾里斑),如右侧的同心圆所示。那么,当 P'、Q' 相距太近的时候,两个斑会叠加导致难以分辨。这就要求物体上的 P、Q 要相距一定的距离。
1873 年,德国物理学家、卡尔蔡司公司的恩斯特·阿贝(Ernst Abbe)首次推算出衍射导致的分辨率极限。根据瑞利判据——“当一个像斑的中心落到另一个像斑的边缘时,就算这两个像刚好能被分辨”,显微镜能分辨的物体上两点 P、Q 的最小距离 h 为:

这个公式就是光学显微镜的分辨率公式,或称为光学衍射极限。(注意此处的分辨率与通常说的显示器分辨率含义不同)
其中, 为光的波长,n 为物方的折射率,
为光的波长,n 为物方的折射率, 为物体与物镜边缘连线和光轴的夹角。如上图所示。为方便起见,其中的
为物体与物镜边缘连线和光轴的夹角。如上图所示。为方便起见,其中的  通常称为数值孔径,简写为 NA。
通常称为数值孔径,简写为 NA。
摄影领域常用 f/# 或称光圈值来描述镜头,光圈值与数值孔径可以相互换算。对于一枚光圈为 2.0 的镜头来说,数值孔径为 0.25,其分辨能力约 1.2 微米。
目前常用的高倍物镜 NA 最大为1.49,可以算出,对于可见光,比如波长 500 纳米的绿光,显微镜的分辨率约为 200 纳米。所以,即使再提高物镜的放大倍率,也不能提高显微镜的分辨率。
而 200 纳米这个数值也就通常被称作衍射极限。
2.5. 光学衍射极限的动态范围
从上面的公式可以看到,光学衍射极限其实并不是一个具体的数值,如果改变上述公式中的参数,是可以有效提高分辨率的。
(1) 光的波长
可见光波长范围是400~760 nm,如果使用更短波长的光,比如紫外线,理论上可以提高分辨率。但是紫外线能量高,易损伤样品,而且透射能力低,很难透过物镜。人们想到了使用高能电子束代替光束,比如 200 keV 的电子对应的的布罗意波长为 0.0025 纳米 (2.5 * 米)。虽然 NA 相对较小(约为10°),依然可以达到0.1 纳米的理论分辨率。这就是电子显微镜的基本原理。
米)。虽然 NA 相对较小(约为10°),依然可以达到0.1 纳米的理论分辨率。这就是电子显微镜的基本原理。
严格的说,电镜的分辨率依然限制在光学衍射极限的范围内。只不过这里的“光学”是“电子光学”。
(2) 物方折射率 n
空气折射率为 1,水的折射率 1.33,玻璃折射率 1.58。目前主要的物镜都是玻璃材质,并在物镜与样品之间用与玻璃折射率一致的油来浸润,以提高分辨率。
2012 年 Olympus 发布了一款 NA 高达 1.7 的物镜,光学部分使用蓝宝石(折射率约 1.76)制作,并搭配高折射率的镜油(目测成分应该是二碘甲烷 Methylene iodide)。
也许在未来能发明比玻璃更好的材料,折射率更高、易于制作透镜、并且能找到高折射率的油,这样就能进一步提高分辨率。
比如用钻石(折射率大约2.42)打造一枚土豪物镜,并找到同样折射率的透明液体,分辨率可以提高到 1.5 倍。当然,由于成本及工艺因素,目前尚不现实。
(3) 孔径角
看起来 的最大值是 90°,但是如果在样品的上下两面都放置物镜,相机同时收集这两个物镜中的光呢?
这种方法就是 Stephen Hell 在 1991 年发明的 4 Pi 显微技术。此时衍射极限的公式稍有变化,分辨率能提高一倍。
简单理解,限制分辨率的一个原因是从物体上向四面八方发射的光线在经过透镜时,由于透镜大小(孔径)的限制,所以很多光线没有经过物体,其承载的物体的“信息”丢失了。而 4 Pi 其实是通过两个物镜收集了更多的光及“信息”。每个高 NA 的物镜离物体都非常近,所以几乎能收集到各个角度的光,这也是 4 Pi 这个名称的来历——一个以物体为球心的球体其立体角的大小就是 。
。
另一种“结构照明显微技术(SIM)”很聪明地通过类似摩尔纹的原理,获取到更多“信息”,也可以将分辨率提高一倍。解释原理需要用到傅里叶光学,在此就不详述了。它由已故的 Mats Gustafsson 教授于 2000 年发明。
那么问题来了,是否还有办法“真正”突破衍射极限,使之并不受限于这个公式呢?
3. 突破光学衍射极限
前面的公式推导过程中,有两个隐含条件。
第一个隐含条件是需要用到夫琅禾费衍射,而它也是有条件的,它要求物体与像平面之间的距离远远大于光的波长。这个条件称为远场条件。如果离物体足够近,衍射极限便不遵循上述公式。研究波长以内光传播特性的领域叫近场光学。
Eric Betzig 在 1986 年发明的近场扫描光学显微技术(NSOM)就是使用距离物体远小于波长的光学探针来扫描物体,以达到超分辨的显微图像。
4. 单分子荧光与分辨率革命(revolution of resolution)
另一个条件则非常之隐蔽,但却十分朴素,以至于自阿贝推算出衍射极限公式之后的 100 多年间,没有人能够发现或重视。
Stefan Hell 与 Eric Betzig 的贡献就在于解锁了这个隐藏关卡,在固定的波长与 NA 之下,将显微镜的理论分辨能力提高到无限大,带来了 1990 年代以来的分辨率革命。而这次革命非常依赖于 William Moerner 推动的单分子荧光成像等研究,也是为什么本次诺奖会颁发给几乎没做过超分辨显微技术的 William Moerner、颁发给理综奖的原因。
这里要简单插播一下荧光成像的原理:荧光分子(如获得 2008 年的诺贝尔理综奖的绿色荧光蛋白 GFP)在吸收一个高能量的短波长(如蓝光,称为激发光)跃迁到高能态之后,很快会发射一个低能量的长波长(如绿光,称为发射光)光子回到基态能级。
所以我们可以用蓝光照射样品上标记的 GFP,而接收其发射的绿光,来对物体进行成像。这样的一个好处就是图像对比度会非常高,只有带有 GFP 的部位会被成像,其它部位都是暗的。
而每个荧光分子的发光是完全独立的。
回到前面说的第二个隐藏条件,说穿了其实非常简单。推导时假定了图 1 中的物体上的两点 P、Q 同时发光,所以二者的衍射斑才会叠加在一起导致难以区分。但如果 P、Q 是在不同的时间分别发光,那么可以(通过点扩散函数拟合或者去卷积的方法)精确地定位到每个衍射斑的中心点位置 P'、Q'。那么也就不存在这样的衍射极限了,理论上即使 P、Q 相距再近都可以被分别定位而加以区分。
更进一步,广义地来说,只要是 P、Q 两点处在可以探测到的不同“状态”或是不同特征,那么就可以超越衍射极限。分辨率并不仅仅是关于波的,而且是关于“状态”的!
意识到了这一点,很多种真正突破衍射极限的显微技术就被解锁了。比如:
(1) 区分荧光分子的不同构象/能态
荧光分子处在不同构象或能态时,可以发射不同波长的光子,或者不发光。
受激发射耗损显微术(STED):激发光周围套上一圈耗损光,使中心区域的荧光分子处于激发态发光,周围的分子处于耗损态不发光。这样可以使图 1 中的同心圆表示的衍射斑变小,以此来区分两个相距在衍射极限以内的分子。这个方法由 Stefan Hell 在 1994 年发明。
随机光学重构显微术(STORM)、光活化定位显微术(PALM)、荧光活化定位显微术(fPALM):它们的共同特征是使样品中每次仅有少量随机、离散的单个荧光分子发光,通过拟合,找出每个荧光分子中心点的位置。重复拍摄多张图片之后,就可以把所有荧光分子的中心点位置叠加起来形成整幅图像。这些技术由庄小威、Eric Betzig 等人在 2006 分别独立发明。

图 2. 庄小威发明的 STORM 超分辨光学成像效果图。a, b 为传统光学显微镜拍摄,c, d 为相同区域经 STORM 技术拍摄并重构。b, d 为 a, c 的局部放大。图中的结构是细胞内的支架——微管蛋白。图片来自 Nikon MicroscopyU 。
RESOLFT:由于 STED 中,耗损光需要很强的亮度,激光器造价昂贵,而且对样品损伤较大,如果借鉴 STORM/PALM/fPALM 的原理,不需要使周围分子处于“耗损态”,而是采用类似于 STORM 等的特殊荧光分子,用较弱的光强使其处于其它能态,也可以实现超分辨。这也是 Stefan Hell 发明的。
(2) 荧光分子闪烁的不同周期
SOFI:有些荧光分子在持续的激发光照射下,并不是持续发光,而是会闪烁。由于每个荧光分子随机闪烁的时间不一样,可以通过连续拍摄一段录像,分析比较图像中每个像素点记录到的荧光强度在这段时间内的闪烁情况,来区分不同的荧光分子,实现超分辨。2009 年发明。
(3) 荧光分子的极性/朝向不同
SPoD/ExPAN:荧光分子都是偶极子,可以理解成有着不同的朝向。当用不同朝向的偏振光激发时,荧光分子的亮度也会随之不同。可以周期性地改变偏振光朝向,连续拍摄一段录像,分析比较图像中每个像素点荧光强度的周期变化情况,来区分不同荧光分子,实现超分辨。2014 年发明。
……
这些不同的方法,也都有着各自分辨率的计算公式。STED/RESOLFT 的理论分辨率与激发光、耗损光强度有关;STORM/PALM 与单个荧光分子发射的光子数有光;SOFI/SPoD 与相机的像素尺寸有关(采样定理决定)。在固定的波长、NA 下,理论上的分辨率依然可以达到任意高。
如果还能想到荧光分子的更多特性并加以利用,未来还会有越来越多的超分辨显微术被发明。
所以可以看到,William Moerner 开创的单分子荧光成像在超分辨成像中的作用。Stefan Hell、Eric Betzig 使用物理方法利用了荧光分子的化学特性而发明的超分辨显微成像,目前在生物学领域已经得到了广泛应用,因此,给三人颁发一个诺贝尔理综奖也是名至实归的。
------------------------
有兴趣了解超分辨实现方法细节的同学,推荐 @Hydro Ding 的这篇回答,比我写的清楚:光学显微镜受光线的波长限制只能够放大两千倍左右的极限,有办法解决么?或者这就是自然定律的一种界限表现? 显示全部
------------------------------------------------
2014 年的诺贝尔理综奖颁发给了“超分辨荧光显微技术”。也许接下来的几天,媒体会关注 Stefan Hell、Eric Betzig 二人的传奇经历,或者另一名华人女科学家与该奖项失之交臂的遗憾。但是八卦之外,这项成果背后的科学本身也非常有意思。
这里面有三个关键词:“超分辨”、“荧光”和“显微技术”,我希望能够解释清楚以下几个问题,尤其是后两个问题:
1. 为什么需要(光学)显微技术?
2. 为什么光学显微镜的分辨率存在理论极限?
3. 用怎样的方法可以突破这个理论极限以达到“超分辨”?为什么这个理论极限可以被突破?
4. 为什么非得是荧光显微技术,而非普通的明场(透射光)显微技术?
1. 采样定理与显微镜
我们用肉眼观察或者用相机拍摄一个物体时,物体上的每一个细微的点都会在眼睛的视网膜或是相机的感光芯片上成像。那么我们为什么不能看到细菌等微小的东西,为什么不能把照片无限放大以看清远处树木上面的每一片叶子呢?
这个问题的答案比较简单:因为组成视网膜的每一个感光细胞(视杆细胞和视锥细胞)、相机芯片上的每一个感光元件(CCD、CMOS等)都是有大小的。比如视网膜中央凹区域的视锥细胞直径平均约为 5 微米。而由于奈奎斯特-香农采样定理的限制,视网膜上能分清的两个相邻像点的距离是视锥细胞直径的两倍,即 10 微米。再结合眼球的构造,大致可以推断出,在距离眼睛 25 厘米的位置,我们能分辨物体上相距为 80 微米的两个点,换算成点阵密度就是大约 320 ppi,这也是苹果所谓“视网膜屏”分辨率的来历。
如果要观察小于 80 微米的物体,比如细菌,就需要先将物体放大,再用眼睛或者相机观察。现代光学显微镜的构造其实非常简单,样品放置在物镜的焦点处,从样品上发射或散射的光经过物镜变成平行(准直)光,再经过一个结像透镜,然后会聚到相机的感光芯片上成像。
按照前面的方法来推算,要区分物体上相距为 200 纳米的两个点,如果使用科研级相机,比如最近火起来的 sCMOS 相机(每个感光像素尺寸为 6.5 微米),只需要使用放大倍率为 65 倍的物镜就足够了。
那么是否可以通过提高物镜的放大倍率来观察低于 200 纳米的物体,比如细胞里面微管呢?
答案是不可以。
2. 光学衍射极限
由于光是一种电磁波,具有衍射和干涉的特性。
图 1. 光学显微镜简化示意图如上面的简图所示,紫色箭头表示的物体 PQ 经过物镜等之后在相机上成像为P'Q'。由于光的衍射,物体上的点如 P、Q,在相机上并不是单独的点,而是一个个有一定大小的斑,被称为夫琅禾费衍射斑(或称艾里斑),如右侧的同心圆所示。那么,当 P'、Q' 相距太近的时候,两个斑会叠加导致难以分辨。这就要求物体上的 P、Q 要相距一定的距离。
1873 年,德国物理学家、卡尔蔡司公司的恩斯特·阿贝(Ernst Abbe)首次推算出衍射导致的分辨率极限。根据瑞利判据——“当一个像斑的中心落到另一个像斑的边缘时,就算这两个像刚好能被分辨”,显微镜能分辨的物体上两点 P、Q 的最小距离 h 为:
这个公式就是光学显微镜的分辨率公式,或称为光学衍射极限。(注意此处的分辨率与通常说的显示器分辨率含义不同)
其中,
摄影领域常用 f/# 或称光圈值来描述镜头,光圈值与数值孔径可以相互换算。对于一枚光圈为 2.0 的镜头来说,数值孔径为 0.25,其分辨能力约 1.2 微米。
目前常用的高倍物镜 NA 最大为1.49,可以算出,对于可见光,比如波长 500 纳米的绿光,显微镜的分辨率约为 200 纳米。所以,即使再提高物镜的放大倍率,也不能提高显微镜的分辨率。
而 200 纳米这个数值也就通常被称作衍射极限。
2.5. 光学衍射极限的动态范围
从上面的公式可以看到,光学衍射极限其实并不是一个具体的数值,如果改变上述公式中的参数,是可以有效提高分辨率的。
(1) 光的波长
可见光波长范围是400~760 nm,如果使用更短波长的光,比如紫外线,理论上可以提高分辨率。但是紫外线能量高,易损伤样品,而且透射能力低,很难透过物镜。人们想到了使用高能电子束代替光束,比如 200 keV 的电子对应的的布罗意波长为 0.0025 纳米 (2.5 *
严格的说,电镜的分辨率依然限制在光学衍射极限的范围内。只不过这里的“光学”是“电子光学”。
(2) 物方折射率 n
空气折射率为 1,水的折射率 1.33,玻璃折射率 1.58。目前主要的物镜都是玻璃材质,并在物镜与样品之间用与玻璃折射率一致的油来浸润,以提高分辨率。
2012 年 Olympus 发布了一款 NA 高达 1.7 的物镜,光学部分使用蓝宝石(折射率约 1.76)制作,并搭配高折射率的镜油(目测成分应该是二碘甲烷 Methylene iodide)。
也许在未来能发明比玻璃更好的材料,折射率更高、易于制作透镜、并且能找到高折射率的油,这样就能进一步提高分辨率。
比如用钻石(折射率大约2.42)打造一枚土豪物镜,并找到同样折射率的透明液体,分辨率可以提高到 1.5 倍。当然,由于成本及工艺因素,目前尚不现实。
(3) 孔径角
看起来
这种方法就是 Stephen Hell 在 1991 年发明的 4 Pi 显微技术。此时衍射极限的公式稍有变化,分辨率能提高一倍。
简单理解,限制分辨率的一个原因是从物体上向四面八方发射的光线在经过透镜时,由于透镜大小(孔径)的限制,所以很多光线没有经过物体,其承载的物体的“信息”丢失了。而 4 Pi 其实是通过两个物镜收集了更多的光及“信息”。每个高 NA 的物镜离物体都非常近,所以几乎能收集到各个角度的光,这也是 4 Pi 这个名称的来历——一个以物体为球心的球体其立体角的大小就是
另一种“结构照明显微技术(SIM)”很聪明地通过类似摩尔纹的原理,获取到更多“信息”,也可以将分辨率提高一倍。解释原理需要用到傅里叶光学,在此就不详述了。它由已故的 Mats Gustafsson 教授于 2000 年发明。
那么问题来了,是否还有办法“真正”突破衍射极限,使之并不受限于这个公式呢?
3. 突破光学衍射极限
前面的公式推导过程中,有两个隐含条件。
第一个隐含条件是需要用到夫琅禾费衍射,而它也是有条件的,它要求物体与像平面之间的距离远远大于光的波长。这个条件称为远场条件。如果离物体足够近,衍射极限便不遵循上述公式。研究波长以内光传播特性的领域叫近场光学。
Eric Betzig 在 1986 年发明的近场扫描光学显微技术(NSOM)就是使用距离物体远小于波长的光学探针来扫描物体,以达到超分辨的显微图像。
4. 单分子荧光与分辨率革命(revolution of resolution)
另一个条件则非常之隐蔽,但却十分朴素,以至于自阿贝推算出衍射极限公式之后的 100 多年间,没有人能够发现或重视。
Stefan Hell 与 Eric Betzig 的贡献就在于解锁了这个隐藏关卡,在固定的波长与 NA 之下,将显微镜的理论分辨能力提高到无限大,带来了 1990 年代以来的分辨率革命。而这次革命非常依赖于 William Moerner 推动的单分子荧光成像等研究,也是为什么本次诺奖会颁发给几乎没做过超分辨显微技术的 William Moerner、颁发给理综奖的原因。
这里要简单插播一下荧光成像的原理:荧光分子(如获得 2008 年的诺贝尔理综奖的绿色荧光蛋白 GFP)在吸收一个高能量的短波长(如蓝光,称为激发光)跃迁到高能态之后,很快会发射一个低能量的长波长(如绿光,称为发射光)光子回到基态能级。
所以我们可以用蓝光照射样品上标记的 GFP,而接收其发射的绿光,来对物体进行成像。这样的一个好处就是图像对比度会非常高,只有带有 GFP 的部位会被成像,其它部位都是暗的。
而每个荧光分子的发光是完全独立的。
回到前面说的第二个隐藏条件,说穿了其实非常简单。推导时假定了图 1 中的物体上的两点 P、Q 同时发光,所以二者的衍射斑才会叠加在一起导致难以区分。但如果 P、Q 是在不同的时间分别发光,那么可以(通过点扩散函数拟合或者去卷积的方法)精确地定位到每个衍射斑的中心点位置 P'、Q'。那么也就不存在这样的衍射极限了,理论上即使 P、Q 相距再近都可以被分别定位而加以区分。
更进一步,广义地来说,只要是 P、Q 两点处在可以探测到的不同“状态”或是不同特征,那么就可以超越衍射极限。分辨率并不仅仅是关于波的,而且是关于“状态”的!
"Resolution is about waves and states." -- by Stefan Hell
意识到了这一点,很多种真正突破衍射极限的显微技术就被解锁了。比如:
(1) 区分荧光分子的不同构象/能态
荧光分子处在不同构象或能态时,可以发射不同波长的光子,或者不发光。
受激发射耗损显微术(STED):激发光周围套上一圈耗损光,使中心区域的荧光分子处于激发态发光,周围的分子处于耗损态不发光。这样可以使图 1 中的同心圆表示的衍射斑变小,以此来区分两个相距在衍射极限以内的分子。这个方法由 Stefan Hell 在 1994 年发明。
随机光学重构显微术(STORM)、光活化定位显微术(PALM)、荧光活化定位显微术(fPALM):它们的共同特征是使样品中每次仅有少量随机、离散的单个荧光分子发光,通过拟合,找出每个荧光分子中心点的位置。重复拍摄多张图片之后,就可以把所有荧光分子的中心点位置叠加起来形成整幅图像。这些技术由庄小威、Eric Betzig 等人在 2006 分别独立发明。
图 2. 庄小威发明的 STORM 超分辨光学成像效果图。a, b 为传统光学显微镜拍摄,c, d 为相同区域经 STORM 技术拍摄并重构。b, d 为 a, c 的局部放大。图中的结构是细胞内的支架——微管蛋白。图片来自 Nikon MicroscopyU 。
RESOLFT:由于 STED 中,耗损光需要很强的亮度,激光器造价昂贵,而且对样品损伤较大,如果借鉴 STORM/PALM/fPALM 的原理,不需要使周围分子处于“耗损态”,而是采用类似于 STORM 等的特殊荧光分子,用较弱的光强使其处于其它能态,也可以实现超分辨。这也是 Stefan Hell 发明的。
(2) 荧光分子闪烁的不同周期
SOFI:有些荧光分子在持续的激发光照射下,并不是持续发光,而是会闪烁。由于每个荧光分子随机闪烁的时间不一样,可以通过连续拍摄一段录像,分析比较图像中每个像素点记录到的荧光强度在这段时间内的闪烁情况,来区分不同的荧光分子,实现超分辨。2009 年发明。
(3) 荧光分子的极性/朝向不同
SPoD/ExPAN:荧光分子都是偶极子,可以理解成有着不同的朝向。当用不同朝向的偏振光激发时,荧光分子的亮度也会随之不同。可以周期性地改变偏振光朝向,连续拍摄一段录像,分析比较图像中每个像素点荧光强度的周期变化情况,来区分不同荧光分子,实现超分辨。2014 年发明。
……
这些不同的方法,也都有着各自分辨率的计算公式。STED/RESOLFT 的理论分辨率与激发光、耗损光强度有关;STORM/PALM 与单个荧光分子发射的光子数有光;SOFI/SPoD 与相机的像素尺寸有关(采样定理决定)。在固定的波长、NA 下,理论上的分辨率依然可以达到任意高。
如果还能想到荧光分子的更多特性并加以利用,未来还会有越来越多的超分辨显微术被发明。
所以可以看到,William Moerner 开创的单分子荧光成像在超分辨成像中的作用。Stefan Hell、Eric Betzig 使用物理方法利用了荧光分子的化学特性而发明的超分辨显微成像,目前在生物学领域已经得到了广泛应用,因此,给三人颁发一个诺贝尔理综奖也是名至实归的。
------------------------
有兴趣了解超分辨实现方法细节的同学,推荐 @Hydro Ding 的这篇回答,比我写的清楚:光学显微镜受光线的波长限制只能够放大两千倍左右的极限,有办法解决么?或者这就是自然定律的一种界限表现? 显示全部
泻药,睡前简单占个坑,明天去请教楼下的做STED的组。
首先是衍射极限,由于光的波动性,普通光学仪器的分辨率受限与工作波长和数值孔径,大约在为光的波长量级。聚焦光斑点点扩散函数(PSF)描述了系统的分辨率。
荧光是光致发光效应的一种,在电子受激进入不同的电子单重态S2后,通过无辐射的系内转换等进入能量稍低的第一激发单重态S1中,再通过振动弛豫的方式到达S1中能量最低的振动能级上,然后回到基态的振动能级中并发射出光子。
如下图Jablonski 能图所示 早期采用荧光技术的有共焦、多光子显微镜,采用荧光染料对特定的组织细胞进行染,然后用激光激发产生荧光,大大提高了样品亮度。
早期采用荧光技术的有共焦、多光子显微镜,采用荧光染料对特定的组织细胞进行染,然后用激光激发产生荧光,大大提高了样品亮度。
摘自楼下某老师的主页
超分辨光学显微实验室(Superresolution Optical Microscopy Lab)
ZEISS Microscopy Online Campus
目前世界领先(就是这次获奖的Hell小组)的分辨率已经在纳米级别了。比微米级别的共聚焦、多光子、OCT等高了几个数量级。
应用场景和改变等,应该让实际使用者等人来说。。。窝就抛砖引玉吧。。至于为什么化学奖 。。。。
午睡起来后给排名第一的答案补充一些光学的东西。 显示全部
首先是衍射极限,由于光的波动性,普通光学仪器的分辨率受限与工作波长和数值孔径,大约在为光的波长量级。聚焦光斑点点扩散函数(PSF)描述了系统的分辨率。
荧光是光致发光效应的一种,在电子受激进入不同的电子单重态S2后,通过无辐射的系内转换等进入能量稍低的第一激发单重态S1中,再通过振动弛豫的方式到达S1中能量最低的振动能级上,然后回到基态的振动能级中并发射出光子。
如下图Jablonski 能图所示
早期采用荧光技术的有共焦、多光子显微镜,采用荧光染料对特定的组织细胞进行染,然后用激光激发产生荧光,大大提高了样品亮度。摘自楼下某老师的主页
超分辨光学显微实验室(Superresolution Optical Microscopy Lab)
ZEISS Microscopy Online Campus
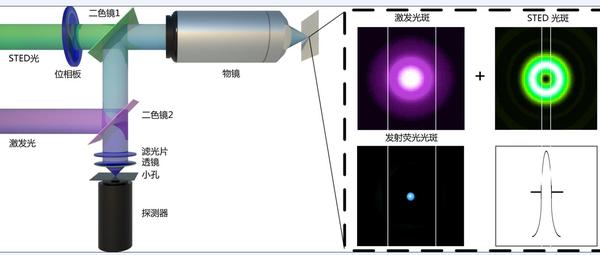
激发射损耗显微镜(STED)其基本原理如下图所示, 紫色代表的是激发激光,绿色代表的是用来受激发射损耗的激光,两束激光经过时间空间调制后同时照射在样本上。激发光使荧光物质发光的同时,STED激光发射一束紧挨着的、环型的、波长较长的激光将激发光斑中大部分的荧光物质通过光学非线性作用被强行回到基态抑制其发荧光,从而减少荧光发射光斑面积。激发光光斑经STED激光的调制后大大的减少了发射荧光分子的光斑大小,并且随着STED激光光强的增加,能发射荧光光斑越小,其半高全宽可以达到衍射极限以下,分辨率不再受光的衍射所限制,从而打破衍射极限。通过三维扫描可以得到50nm以下的三维图像。
目前世界领先(就是这次获奖的Hell小组)的分辨率已经在纳米级别了。比微米级别的共聚焦、多光子、OCT等高了几个数量级。
应用场景和改变等,应该让实际使用者等人来说。。。窝就抛砖引玉吧。。至于为什么化学奖 。。。。
午睡起来后给排名第一的答案补充一些光学的东西。 显示全部

占坑。慢慢更新答案。先贴上ZEISS公司做的很棒的一个关于超分辨的介绍。ZEISS Microscopy Online Campus
排名第一的答案已经很清楚详细,但是对于外行而言这种行文可读性还是较差。所以我来补充一个答案吧,只需要有基础的物理常识就能看懂。
STED的原理和PALM或者STORM极为不同,这边介绍PALM和STORM主要概念吧(具体工作原理不表),保证简单易懂。
任何不论多小的光点发光,由于衍射的关系,被观测到的都是一坨光斑,几纳米大小的发光颗粒被观测到的其实是一个直径几百纳米的一个圆形光斑,所以如果有两个发光颗粒它们之间距离在几十到上百纳米左右,那么我们是无法分辨出到底是一个发光点还是两个的,因为看起来都是一坨嘛,也就是说普通光学成像分辨率就在几百纳米。
就像这样:
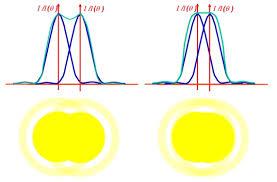 右边太近的两个光点你就分不清了啊。
右边太近的两个光点你就分不清了啊。
这对于观测纳米大小的分子的具体位置很不管用,这让人感到难过对吧。
但是,前文说了,单个发光点发出的光看起来是圆形的一坨,那么如果我告诉你,你看到的那坨光里面确定只有一个发光点的话,你能不能猜出发光点具体在哪里?当然能!废话嘛,就在那个圆斑的中心!
好了, 话到这里你其实已经理解什么叫超分辨率成像了。
但问题又来了,你平时看东西的时候看到的画面都是无数个发光点一起发光构成的。你根本没办法让你的观测物单点单点的发光给你看啊。超分辨成像技术的核心就是解决了这个问题。
得简单介绍下荧光成像才能继续往下编故事了。
荧光,不是你去演唱会玩的荧光棒啊。那是一种分子发光机制,简单说,就是吸收一个高能量的光子,再放出一个低能量的光子。这个技术在生物学特别好用。原因是,生物学家有办法让生物表达能够发荧光的蛋白质!或者把其它的荧光分子弄到生物体内。那么如果一只老鼠体的神经细胞里有绿色荧光蛋白,那么用蓝光照一下,就能看到它神经细胞上发着绿油油荧光的一个个点(蓝光能量比绿光高)。
像这样:
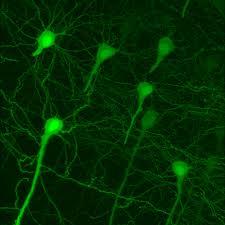
荧光成像,发光的不是连续的一片光源,而是散开的一个个小光点。 光点足够多的话就能看清楚生物结构了呢,超好用!但是它也有局限,就是由于荧光发光点密集,所以能够看到的那些细丝最细也得几百纳米!而生物结构精细处只有几纳米啊,我就是要看清楚该怎么办?
最后一步高能预警
如果,我告诉你,上图中所有光点都能逐一亮起来,每亮起一个你看到一个几百纳米的圆斑,你去找到它的中心(实际操作中可以精确到几纳米),然后把所有亮斑的中心位置再次描到上面这张图里,那你不就能看到几微米的细微结构了嘛?!!!
PALM和STORM不用管它是怎么做到的,核心就是让荧光分子不同时发光,可以让你逐一去做中心定位!!!
于是你就能看到更加清晰的图像啦,偶也!
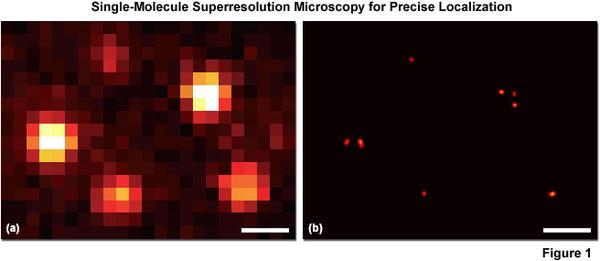 左边是中心定位前,右边是中心定位后。分辨率提升了1-2个数量级啊!
左边是中心定位前,右边是中心定位后。分辨率提升了1-2个数量级啊!
就是这样。
==========================================================================
补充说一下STED吧,它的原理也不表了,核心就是让上图左面的那张里的单分子光斑本身变小,你就连找中心这步都不用了,因为它本身已经足够小了,分辨率就很高了。它不需要让荧光分子逐一亮起。 显示全部
排名第一的答案已经很清楚详细,但是对于外行而言这种行文可读性还是较差。所以我来补充一个答案吧,只需要有基础的物理常识就能看懂。
STED的原理和PALM或者STORM极为不同,这边介绍PALM和STORM主要概念吧(具体工作原理不表),保证简单易懂。
任何不论多小的光点发光,由于衍射的关系,被观测到的都是一坨光斑,几纳米大小的发光颗粒被观测到的其实是一个直径几百纳米的一个圆形光斑,所以如果有两个发光颗粒它们之间距离在几十到上百纳米左右,那么我们是无法分辨出到底是一个发光点还是两个的,因为看起来都是一坨嘛,也就是说普通光学成像分辨率就在几百纳米。
就像这样:
右边太近的两个光点你就分不清了啊。这对于观测纳米大小的分子的具体位置很不管用,这让人感到难过对吧。
但是,前文说了,单个发光点发出的光看起来是圆形的一坨,那么如果我告诉你,你看到的那坨光里面确定只有一个发光点的话,你能不能猜出发光点具体在哪里?当然能!废话嘛,就在那个圆斑的中心!
好了, 话到这里你其实已经理解什么叫超分辨率成像了。
但问题又来了,你平时看东西的时候看到的画面都是无数个发光点一起发光构成的。你根本没办法让你的观测物单点单点的发光给你看啊。超分辨成像技术的核心就是解决了这个问题。
得简单介绍下荧光成像才能继续往下编故事了。
荧光,不是你去演唱会玩的荧光棒啊。那是一种分子发光机制,简单说,就是吸收一个高能量的光子,再放出一个低能量的光子。这个技术在生物学特别好用。原因是,生物学家有办法让生物表达能够发荧光的蛋白质!或者把其它的荧光分子弄到生物体内。那么如果一只老鼠体的神经细胞里有绿色荧光蛋白,那么用蓝光照一下,就能看到它神经细胞上发着绿油油荧光的一个个点(蓝光能量比绿光高)。
像这样:
荧光成像,发光的不是连续的一片光源,而是散开的一个个小光点。 光点足够多的话就能看清楚生物结构了呢,超好用!但是它也有局限,就是由于荧光发光点密集,所以能够看到的那些细丝最细也得几百纳米!而生物结构精细处只有几纳米啊,我就是要看清楚该怎么办?
最后一步高能预警
如果,我告诉你,上图中所有光点都能逐一亮起来,每亮起一个你看到一个几百纳米的圆斑,你去找到它的中心(实际操作中可以精确到几纳米),然后把所有亮斑的中心位置再次描到上面这张图里,那你不就能看到几微米的细微结构了嘛?!!!
PALM和STORM不用管它是怎么做到的,核心就是让荧光分子不同时发光,可以让你逐一去做中心定位!!!
于是你就能看到更加清晰的图像啦,偶也!
左边是中心定位前,右边是中心定位后。分辨率提升了1-2个数量级啊!就是这样。
==========================================================================
补充说一下STED吧,它的原理也不表了,核心就是让上图左面的那张里的单分子光斑本身变小,你就连找中心这步都不用了,因为它本身已经足够小了,分辨率就很高了。它不需要让荧光分子逐一亮起。 显示全部
 知乎用户,专注电磁隐身衣,酷爱英伦摇滚
知乎用户,专注电磁隐身衣,酷爱英伦摇滚
收起
知乎用户 赞同
看了得票最高的回答,觉得很有意思,也很受启发。
一般定义的衍射极限称之为abbe极限,值得注意的是,它主要针对的是光学显微镜的分辨率。
上次听了一个南洋理工大学教授的报告,在报告上大家就这个衍射极限作了一些深入的讨论。这个教授做的是用电磁波逆问题的思路来成像,简单来说,就是先测量一个物体周围的场,然后通过一个数据处理来确定这个物体的形状及电磁参数。
在讨论衍射极限时,他提出光学显微镜这个物理过程,实质上就是一个数据处理过程(data processing),详细说应该是一个逆傅里叶变换。完美的逆傅里叶变换是可以完美得到物体信息的,即完美透镜。完美透镜首先是由J.B. Pendry, 提出来的,这个透镜的电磁参数是磁导率为-1,介电常数也为-1,即负折射率材料。如果不是完美透镜,信息就会丢失,比如光学显微镜采用的透镜就不是完美的,所以丢失很多信息不能达到完美成像,即导致了衍射极限的出现。
在完美透镜与传统透镜之间还存在一些透镜,比如双曲线色散的材料做成的透镜,即超透镜(superlens),这些透镜一般都很容易突破衍射极限。
教授在用他的方法成像的时候也很容易突破衍射极限,这是因为他采用的数据处理方法不是逆傅里叶变换,而是另外一种方法,称之为MUSIC(Multiple Signal Classification 多信号分类)。
也就是说如果采用别的数据处理方法,突破abbe极限其实并不是什么问题。
现在再讲讲超分辨荧光显微技术,我以前并不了解这种成像方法,看了最高得票答案才有所了解。不过我觉它也是一种不同于光学显微镜的数据处理方法。在我看来,超分辨荧光显微技术在有个方面优于传统成像技术,那就是它利用了时间维度的信息。
这些是我个人观点,仅供参考。
-------------------------------------------------------------
我在这里对 @袁霖做一个回复。
完美透镜在光频段还没实现,但是superlens或者hyperlens已经实现了。它是这样的:
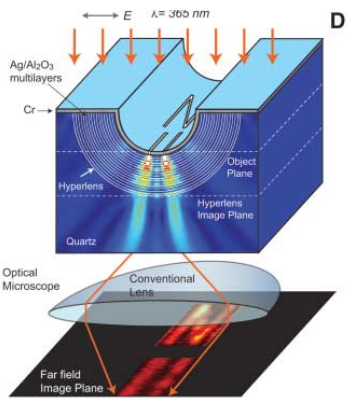
根据文中指出,这个透镜至少达到 的分辨率。
的分辨率。
这种技术确实很有商用潜力,应该也可以用来观察活体。这在文中也有指出:
The optical hyperlens opens up exciting possibilities in applications,such as real-time bio-molecular imaging and nanolithography.
但是我觉得超分辨荧光显微技术可能更加适合观察活体,它利用了活体能够吸收荧光分子的特点。
总的来说,如果超分辨荧光显微技术获诺奖并不是仅仅在于其超高的分辨率,而是在于它在化学或者生物上的巨大的应用价值。这也是为什么其获得的是诺贝尔化学奖,而不是物理学奖。 显示全部
一般定义的衍射极限称之为abbe极限,值得注意的是,它主要针对的是光学显微镜的分辨率。
上次听了一个南洋理工大学教授的报告,在报告上大家就这个衍射极限作了一些深入的讨论。这个教授做的是用电磁波逆问题的思路来成像,简单来说,就是先测量一个物体周围的场,然后通过一个数据处理来确定这个物体的形状及电磁参数。
在讨论衍射极限时,他提出光学显微镜这个物理过程,实质上就是一个数据处理过程(data processing),详细说应该是一个逆傅里叶变换。完美的逆傅里叶变换是可以完美得到物体信息的,即完美透镜。完美透镜首先是由J.B. Pendry, 提出来的,这个透镜的电磁参数是磁导率为-1,介电常数也为-1,即负折射率材料。如果不是完美透镜,信息就会丢失,比如光学显微镜采用的透镜就不是完美的,所以丢失很多信息不能达到完美成像,即导致了衍射极限的出现。
在完美透镜与传统透镜之间还存在一些透镜,比如双曲线色散的材料做成的透镜,即超透镜(superlens),这些透镜一般都很容易突破衍射极限。
教授在用他的方法成像的时候也很容易突破衍射极限,这是因为他采用的数据处理方法不是逆傅里叶变换,而是另外一种方法,称之为MUSIC(Multiple Signal Classification 多信号分类)。
也就是说如果采用别的数据处理方法,突破abbe极限其实并不是什么问题。
现在再讲讲超分辨荧光显微技术,我以前并不了解这种成像方法,看了最高得票答案才有所了解。不过我觉它也是一种不同于光学显微镜的数据处理方法。在我看来,超分辨荧光显微技术在有个方面优于传统成像技术,那就是它利用了时间维度的信息。
这些是我个人观点,仅供参考。
-------------------------------------------------------------
我在这里对 @袁霖做一个回复。
完美透镜在光频段还没实现,但是superlens或者hyperlens已经实现了。它是这样的:
根据文中指出,这个透镜至少达到
这种技术确实很有商用潜力,应该也可以用来观察活体。这在文中也有指出:
The optical hyperlens opens up exciting possibilities in applications,such as real-time bio-molecular imaging and nanolithography.
但是我觉得超分辨荧光显微技术可能更加适合观察活体,它利用了活体能够吸收荧光分子的特点。
总的来说,如果超分辨荧光显微技术获诺奖并不是仅仅在于其超高的分辨率,而是在于它在化学或者生物上的巨大的应用价值。这也是为什么其获得的是诺贝尔化学奖,而不是物理学奖。 显示全部

李玮祥 赞同
参见哈佛大学庄小薇实验室的工作,Zhuang Home。通俗的一句,各类超分辨荧光显微技术的实质并非超越了abbe原理,而是在一定时间范围内(几飞秒-几微秒)连续多次激发的荧光然后用电脑进行变换解析后,得到的一种概率清晰度。大概表达为用时间换分辨率吧。


No comments:
Post a Comment