了解原子軌域與目睹化學鍵
進行單一顆原子的計算首先我們要在週期性的邊界條件之下進行一顆原子的計算,因此我們要用介面來建立模型並造出 CASTEP 的輸入檔。這是本課程第一次使用 CASTEP 來對 Cerius2 所建構出來的模型進行運算,因以下僅就最初步的功能作一步步的介紹。
在 Cerius2 介面中選用 Crystal Builder → Crystal Building ,在 Lattice Parameter 之中填入晶胞邊長,原子間距最好大於 7 個 Angstron,例如我們可以選 a=8、b=8、c=8(單位是 Angstron),晶格向量夾角維持預設值的直角即可。填完後按 Build Crystal 即會在 Model Window 中出現 8 × 8 × 8 Ang. 的晶胞。
點選 Add Atoms,在跳出的選單中打入元素的種類(或從週期表 icon 的選單中挑選);選擇座標的表示方式為相對於 a, b, c 而非 x, y, z ,之後輸入入 0.5 0.5 0.5 之座標值,最後按 Add,則原子會出現在 supercell 的正中心。模型這樣就做好了,若想先存起來,可以從 File → Save Model → 命名(也可按 comment 鈕加註解)之後按 Save 即可。
我們現在己經有模型可以進行量子力學計算了,要使用 CASTEP,先按著 Modules 選單中選 Quantum II(那裏量子物理計算工具),並選 CASTEP 模組,這時你會看到 Run、Analysis、Job Control 等選單,按下 Run (不用擔心按這裏就會不明不白跑起計算來,這只是所謂的 Run 選單,許多進行計算前的設定都在這個選單之內),則跑 CASTEP 之設定的主選單就會跳出,與現在之初步計算比較有關係的有以下幾點:
(一)在 Task 裏的 Single Point Energy 是指原子與晶胞不變動之下作求解波函數的自冾計算,
(二)另外 Basis Set 是指展開波函數所採用之平面波的數目的大小,為了要精確一點可選擇 Medium 或 Fine。
(三)在 File Prefix 提示語左側之欄位填入你要給這個計算取的基本檔名,也就你要給這個計算所取的名字,它必須是合法的 UNIX 檔名,例如可設成“Ne_atom”。
(四)Title 欄可以填寫自己的註解,將來當有很多計算完成的輸出檔寫在一起時,這些註解可能在幫助整理上會很有用。
CASTEP 的功能強大,可作細部設定的項目很多,對於很多我們沒有去設定的參數,Cerius2 介面都能自動給一個合宜的預設值,現在我們可以開始計算原子的波函數計算了,就是在按左上角的 RUN 鈕。左下角的文字終端視窗會出現一些訊息來報告起動的狀況。此時若要看運算進行得如何,可以按下 Job Control 選項來打開 CASTEP Job Control 選單,這裏面有你過去所跑過的計算,剛剛才丟進去的 job 也會在其中,選它並按 UPDATE Status List 則介面會報告此計算現在是 running 還是 completed,若按 Monitor Logfile 則會跳出 CASTEP 文字輸出檔,我們以後會再告訴大家這裏面有什麼細部的訊息可以告訴我們。
分析原子計算的結果
較大的計算可能耗時小數甚至數日,我們不必一直把介面開著,要看跑完了沒有或是分析結果再開啟 Cerius2 的 CASTEP 介面就可以了。分析之前計算結果的操作是:
在 Quantum II 的 CASTEP 模組選單裏,選 Analyze → Files ,則跳出 CASTEP File Analysis 選單,其中上面的視窗中會列出在這個目錄裏所有延伸檔名叫 *.cst 的檔。選 Ne_atom.cst,畫面會跳出前面計算有關的資訊以供查核,如使用的平面波截止動能大小、使用的交換相干近似種類,以及自行註解的 Title 文字等。在 Model 視窗也會跳出該跑完計算後的結構。完成此 File 之選定後,接下來凡是在 Analyze 選項內的功能,都是針對現在所選的這個計算結果,直到另外選擇要 Analyze 的 File 或完全退出 Cerius2 為止。
在本單元中我們主要的興趣是價電子之原子軌域的能階與波函數。從 Analyze → Orbitals 選項中,我們可以看到佔據態的能階,單位是 eV。(若也想看到未佔據態的能階,請見本單最後的補充說明。)波函數是一個複數值函數,它而在每一個空間點上都有實部及虛部兩組函數,所以作圖的呈現比較不容易。在分析介面中提供的功能是看波函數的 norm ,也就是軌域的機率密度。我們會看到如教科書所告訴我們的 s、p、d 對稱性的原子軌域。操作的方法是先點選想看的能階,再按下 Calculate 鈕,就可在 Model Window 中畫出立體的電子雲機率密度等高面(iso-surface),至於等高面的高度值,可在同樣是 Analyze 選項中的 Surfaces 內作改變以取得更清晰的圖像或趨勢,更多 3D 表面的設定值也可以在那裏調整,大家不妨多玩玩看。若要消去等高面,按 Surfaces 選項內的 Delete Surface。
除了三度空間中的等高面外,二維的等高線圖(Countour Plot)也是常用的分析作圖工具,它不像等高面那樣要設定不同的 isosurface level 來觀察,它一次就能給出不同函數值的等高線,只不過每張圖只能選空間中的一個平面來分析。Isosurface 與 cuntour plot 各有所長,前者藉著曲面的包覆延伸來展示整個量子態的分佈範圍;後者以切片上的強弱來表示機率密度在空間縱深中的變化,兩種工具相得益彰。
畫等高線圖的方法;用 Analyze 中 Slices 的功能,按 Create New Slices 即可,它會自動選電荷密度比較高的地方來切片,當然這樣做不一定剛好會是我們想的地方,此時就可善用切片方位的選擇工具來改變切片平面的法向量及其高度,另外,先把 More Editing Option 裏的 Automatic Determine Slice Direction 關掉,在 Model Window 裏以按滑鼠右鍵轉動模型來決定模型的方位後,再按 Set Slice Direction Parallel to Screen 也是一種非常實用的定切片法方向的方法。
雙原子分子與化學鍵
分子的電子雲分佈,在此以 H2、He2 以及 Cl2、CO 兩組四個為範例,一次跑兩個 job 作比較。
首先依前面之單元所介紹過之建構分子模型的方法,準備好將雙原子分子置於週期性晶胞之中,注意所用的晶胞邊長要夠大到距晶胞邊界有大約 4 Angs. 的空間,這樣分子與分子之間才保證有 8 Angs. 或以上的的間距。有了置於晶胞內的獨立今子模型之後,就可以在模組選單的 Quantum II 類裏頭選 CASTEP,並按 Run 選單來設定計算參數。照跑電子結構的標準步驟,在此我們選 Task 為 Single Point Energy(這是因為分子結構己經知道;如果一開始並不知道鍵長,用 Task 工作選項中的 Geometry Optimization 就可以自動一邊計算一邊移動原子的位置,直到系統總能最低而達平衡狀態為止。詳見下一個單元的介紹。)並選好所要用的 Basis Set 精確度(這裏用 Medium 就可以)。再來於 Title 中填入自行註解的說明文字,最後給整個計算取個名字,如 Cl2_mol,就可以按 RUN 按鈕來進行計算。計算的進度可以透過 Job Control 選項內的功能來監控,或是直接在跑計算的目錄底下以文字模式打“ tail -f Cl2_mol.cst ”(要退出合按 Ctrl-C )。
電子結構跑完之後,己經可以觀看這個分子的總電荷密度了。如同前面己經跟大家介紹過的作法,要分析計算完後的結果要先從 Analyze → File 選擇所要分析的文字輸出檔 LOAD進來,像在這裏的例子就是 Cl2_mol.cst。 我們現在要看這個分子的總價電子雲密度分佈的樣子:打開 Density 選單,按 Display 鈕,密度就會以等高面呈現出來,調節等高面值的設定在 Surfaces 裏。若要看某個切面上的電荷密度等高線圖,一樣選用 Slice 的切片工具,再依選項內的功能調整設定,最後再按 Create Slice Plot in Graph Window。我們可以看到兩個原子之間有電子雲連繫在一起,這就是共價鍵。
等高面基本上給出了分子的輪廓,這是因為範例中的分子都是穩定的化合物,其有電子佔據軌域都是全填滿的,且各原子滿足最穩定的八偶律,故在含電子雲密度高的地方,對其他接近而來電子雲也有會排擠現象,因此由密度 isosurface 的反映了分子的形狀與大小。
在己經知道分子輪廓的情況下,我們還能更進一步在這個輪廓的包覆面上視覺化(visualize)其他的物理性質。例如靜電位勢的大小,把它表現在代表分子實體大小範圍之 Density iso surface 時,就呈現出分的那一個比較會有極性而那一部分比較不會。操作方法是:打開 Property Maps,先從 File Types 中選擇所要貼在輪廓面上之物性的檔案種類,在本例中是選 ELECTROSTATIC POTENTIAL,則上面選單會目前目錄下所可以選擇的檔案,這裏的例子就會是 Cl2_mol.cst_esp,點選並按 LOAD 之後,注意最上面的提示會出現 Add Property Cl2_mol.cst_esp 以供確認,這是我們要的因此按下它左邊的圓點鈕,靜電位勢的大小就會以彩色層次的方式貼圖在分子密度輪廓面上,表現出該分子的極性。
請大家針對 H2、He2、Cl2、CO 這四個分子都看看它們的各項上述特性,看看它們的分子形狀、鍵結剖面、極性等各有什麼異同。
獲得未佔據態以及進行鍵級計算
化學鍵是了解原子間相互束縛效應,以及預測分子或固體結構的非常重要而有用的觀念。它並沒有唯一的定義,而是有許多種有用圖象。化學家(及任何人)在不同的場合可以依需求採用最方便者,路易士(Lewes)電子對、價鍵理論、分子軌域理論都是常見常用。鍵結的種類,依傳統固態物理教科書的分類,則有共價鍵、離子鍵、金屬鍵、氫鍵、凡得瓦鍵等,請參閱固態物理教科書(如 Marder,Condensed Matter Physics 第 11 章)。鍵級(Bond Order)
在量子力學層次上探討化學鍵的問題,不外乎當兩個原子接近時,是波函數、電子佔據狀況、系統總能量如何因此而改變,以及將會形成什麼新的穩定態的構形。這裏面共價鍵所涉及電子的共享於兩個原子之間而使系統總能下降,它直接與波函數、佔據情況、能量有關,是最容易作明確分析的一種化學鍵。
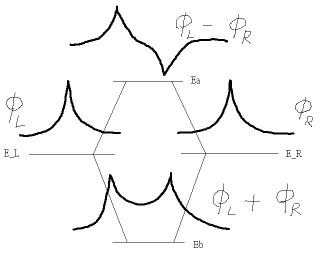
在簡單的 LCAO 近似之下,分子軌域(即分子之單粒子薛丁格方式的解)可以寫成由原子軌域的線性組合而成 φ = cL φL + cR φR ,其中 cL 與 cR 分別為待定的係數(L、R 代表左與右)。在此 LCAO 的近似之下,對於相同原子核的分子,其 bonding 分子軌域是為 φb = φL + φR,而 anti-bonding 分子軌域是為 φa = φL - φR,這是可以被證明的。其中 bonding orbital 的能量較低,而 anti-bonding orbital 的能量則較高。(這主要是因為鍵結電子雲散佈得較開,即波函數曲率小因而動能低;而反鍵的波函數則有節點,故波函數曲率大因而動能高。至於位能的部分則鍵結與反鍵的差異就沒那麼大。)鍵結(Bonding)與反鍵(Anti-Bonding)的一個簡單的圖示如下:
觀看分子的軌域,包含有電子佔據的與空著沒有電子佔據的,對於了解化學鍵(共價鍵)的強弱的確是有很大的助益。在 CASTEP 如何看到這些資訊呢?首先我們必須進行未佔據態的計算(方法不只一種),其中一種方法如下:在完成收斂電子結構與總能的 Single Point Energy 之 Task 後,進入分析模式 Analyze → Density of States 之中,在 DOS Calculation 欄框,按 Setup K-points 鈕,並在跳出之 CASTEP DOS K-points 中填設 Mesh Parameters 為 1 1 1(這是因為單獨分子的量子態不應有多重的 k 值),填後可將選單關閉。再來,改變在 Number of Bands 上的原數值(代表佔據軌域的總數)為加上我們希望要看的未佔據態數目之後和,例如原有 7,而我們希望多看 3 個空軌域,則把 7 改成 10。再勾選 Perform Population Analysis,最後按 Calculate Band Energies 來發動 CASTEP 計算,我們會在 Job Control 選單中看到一個新的計算叫做 Cl2_mol_DOS 正在跑。
上述的計算跑完之後,我們再次用 Analyze → Files(選 Cl2_mol_DOS.cst 來 LOAD)→ Orbitals 來分析佔據及未佔據態的分子軌域。在 CASTEP Orbitals 選單中選擇所要畫的軌域,再按 Calculate 即可顯示。我們會看到具有不同軸對稱的鍵結及反鍵軌域,從從軸心看下去會無節面(nodal plane)、一節面、兩節面的電子雲;它們相當於波爾原子模型角動量是 0、1、2,對原子而言就是 s、p、d,而這也就是 σ、π、δ 鍵命名上的由來。
按基礎化學教科書中的定義,鍵級是被定義為“(鍵結電子數 - 反鍵結電子數)÷2”,像前面的例子 H2 之鍵級是 1,而 He2 鍵級是 0,鍵級代表兩個原子之間其波函數形成共價鍵的程度(離子鍵、氫鍵等就不能使用鍵級來描述了),He2 鍵級是零意味著 He 寧可單原子獨自存在也不喜歡形成分子。國中課本裏有名的乙烷 H3C-CH3、乙烯 H2C=CH2、乙炔 HC≡CH,就是大家熟悉之單、雙、參鍵之例子,其鍵級各為 1、2、3。
鍵級在量子化學分析方法中還有較廣義的定義,可以不受限於以須直接介定一個軌域是鍵結、反鍵結,或是非鍵結(non-bonding)軌域。CASTEP 內建 Muliken Population Analysis,可給出相當於鍵級大小的參考指標(Bond Order Index)。由於我們先前的計算己經選擇了 Perform Population Analysis,因此我們只要去看 CASTEP 的文字輸出檔 Cl2_mol_DOS.cst 就可以看到鍵級指標,查看的方法是,可從 Job Control 裏選這個計算並按 Monitor Log File,或在 UNIX 文字終端機視窗中用 tail、more 或 vi 來看 *.cst 檔。
請大家再次針對 H2、He2、Cl2、CO 這四個分子都看看它們的各項上述特性,看看它們的分子軌域軸向上的對稱性(σ鍵或π鍵)、鍵結與反鍵、以及鍵級指標等各有什麼異同。
No comments:
Post a Comment